
Meldung über produktionsfreie Zeiten oder Produktionszeiten
Start (Copy)
Risk Management for Medical Devices II
Which regulatory requirements must be adhered to minimize risk and which implementation is requested by the MDR? Our auditor and expert, Dr. Hogh-Janovsky, will explain the relevant requirements of the MDR in more detail with you in Part II.
Benennungsurkunde_MDR
Änderungsmitteilung (MDR)
Fragebogen zur Angebotserstellung – Medizinprodukte – Anlage I: Liste Medizinprodukte (MDR)
Preisliste – Zertifizierung nach Verordnung (EU) 2017/745 (MDR)
Basis UDI-DI und deren Vergabe
Die Basis-UDI-DI ist wichtiger Schlüssel in der Produkt-Dokumentation (z. B. Zertifikate, technische Dokumentation, Vigilanz Meldungen und PSUR, SS(C)P, etc.) und dient auch als Zugangsschlüssel zu den produktbezogenen Informationen in der europäischen Datenbank für Medizinprodukte (EUDAMED). Die Basis-UDI-DI stellt dabei eine sinnvolle Möglichkeit dar, mehrere vergleichbare Varianten eines Medizinprodukts zu gruppieren.
Wesentliche Anforderungen für die Festlegung der Basis-UDI-DI sind in MDCG 2018-1 (aktuell Rev. 4) angeführt. Es werden nur Forderungen der 2017/745 (MDR) adressiert, das Dokument wird aber auch für die Verordnung (EU) 2017/746 herangezogen. Diese Anforderungen müssen berücksichtigt werden.
Insbesondere bei der gemeinsamen Gruppierung von Produkten sind wesentliche Aspekte zu berücksichtigen. Die folgenden Datenelemente müssen für alle gemeinsam gruppierten Produkte gleich sein.
Anforderungen für das Zusammenfassen unter derselben Basis-UDI-DI:
- Selber Hersteller (Name, Adresse, SRN)
- Selbe Risikoklasse (MDR: insbesondere hinsichtlich Implantierbarkeit, aktives Medizinprodukt, besondere Bestandteile wie Arzneimittelkomponente, Materialien tierischen Ursprungs oder stoffliche Komponenten sowie Messfunktion oder ob es sich um ein wiederverwendbares chirurgisches Element handelt; IVDR: gemäß Anhang VIII)
- Selber Medical Device Nomenclature Code (EMDN Code) – zumindest bis zur vierten Stelle
- Selbe Zweckbestimmung
- Angeführt auf demselben Produktzertifikat, PSUR, SSCP (MP) bzw. SSP (IVD)
- Gegebenenfalls gleiche (eine) Technische Dokumentation
- Gegebenenfalls s gleiche wesentliche Auslegungs- und Herstellungsmerkmale
Für IVD wird eine Basis-UDI-DI insbesondere für Sprach- und Vertriebsvarianten (z.B. Tests zur Eigenanwendung in verschiedenen Ländern für diverse Distributoren mit länderspezifischer Kennzeichnung) sowie Packungsvarianten des gleichen Produkts (z.B. 96-Well bzw. 128-Well Platten, Mehrfachabpackungen) vergeben.
Insbesondere hinsichtlich derselben wesentlichen Auslegungs- und Herstellungsmerkmale möchten wir für Medizinprodukte auf einige Aspekte hinweisen, welche zu unterschiedlichen Basis-UDI-DI führen:
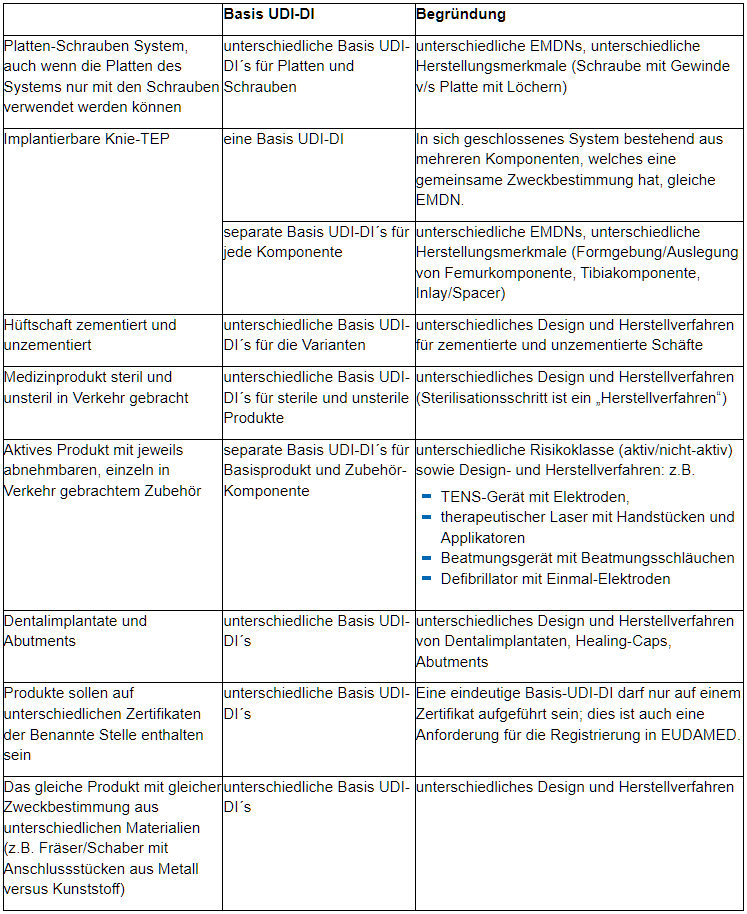
Verbindliche Festlegung der deutschen Marktaufsichtsbehörden (AGMP) zum Umgang mit Konformitätserklärungen für Richtlinien-Produkte (gemäß 93/42/EWG bzw. 98/79/EG)
Zur Zeit besteht keine Notwendigkeit eine aktuelle Konformitätserklärung, wenn es keine Änderungen gibt, ergänzend zur ursprünglichen, vor dem Geltungsbeginn der MDR (26.05.2021) bzw. IVDR (26.05.2022) ausgestellten Konformitätserklärung, mit Bezug auf Art. 120 MDR bzw. Artikel 110 IVDR zu fordern.
Jedoch muss im Falle nicht signifikanter Änderungen, die eine Anpassung der Konformitätserklärung erfordern (z.B. Umfirmierung, Adressänderung) die bestehende Konformitätserklärung gemäß Richtlinie um ein Beiblatt bzw. Anhang ergänzt werden.
Dies wurde nun durch die deutschen Marktaufsichtsbehörden (AGMP) klargestellt und es wird besonders daraufhin gewiesen, dass es KEINE neue Konformitätserklärung nach MDD/ IVDD geben darf, sondern nur ein „Beiblatt“ (s.o.) zu der vor dem 2021-05-26 (MDD)/ 2022-05-26 (IVDD) ausgestellten Konformitätserklärung, welche die nicht signifikanten Änderungen (wie auch neue Produktvarianten) eindeutig nennt.
ISO 9001
Downloads
Struktur Technische Dokumentation (Medizinprodukte)
Technische Dokumentation (In-vitro-Diagnostika) – Dateistruktur (ZIP)
Benennungsurkunde_MDR
Technische Dokumentation (Medizinprodukte) – Dateistruktur (ZIP)
Änderungsmitteilung (MDR)
Fragebogen zur Angebotserstellung – Medizinprodukte – Anlage I: Liste Medizinprodukte (MDR)
Preisliste – Zertifizierung nach Verordnung (EU) 2017/745 (MDR)
Kunden News
Basis UDI-DI und deren Vergabe
Die Basis-UDI-DI ist wichtiger Schlüssel in der Produkt-Dokumentation (z. B. Zertifikate, technische Dokumentation, Vigilanz Meldungen und PSUR, SS(C)P, etc.) und dient auch als Zugangsschlüssel zu den produktbezogenen Informationen in der europäischen Datenbank für Medizinprodukte (EUDAMED). Die Basis-UDI-DI stellt dabei eine sinnvolle Möglichkeit dar, mehrere vergleichbare Varianten eines Medizinprodukts zu gruppieren.
Wesentliche Anforderungen für die Festlegung der Basis-UDI-DI sind in MDCG 2018-1 (aktuell Rev. 4) angeführt. Es werden nur Forderungen der 2017/745 (MDR) adressiert, das Dokument wird aber auch für die Verordnung (EU) 2017/746 herangezogen. Diese Anforderungen müssen berücksichtigt werden.
Insbesondere bei der gemeinsamen Gruppierung von Produkten sind wesentliche Aspekte zu berücksichtigen. Die folgenden Datenelemente müssen für alle gemeinsam gruppierten Produkte gleich sein.
Anforderungen für das Zusammenfassen unter derselben Basis-UDI-DI:
- Selber Hersteller (Name, Adresse, SRN)
- Selbe Risikoklasse (MDR: insbesondere hinsichtlich Implantierbarkeit, aktives Medizinprodukt, besondere Bestandteile wie Arzneimittelkomponente, Materialien tierischen Ursprungs oder stoffliche Komponenten sowie Messfunktion oder ob es sich um ein wiederverwendbares chirurgisches Element handelt; IVDR: gemäß Anhang VIII)
- Selber Medical Device Nomenclature Code (EMDN Code) – zumindest bis zur vierten Stelle
- Selbe Zweckbestimmung
- Angeführt auf demselben Produktzertifikat, PSUR, SSCP (MP) bzw. SSP (IVD)
- Gegebenenfalls gleiche (eine) Technische Dokumentation
- Gegebenenfalls s gleiche wesentliche Auslegungs- und Herstellungsmerkmale
Für IVD wird eine Basis-UDI-DI insbesondere für Sprach- und Vertriebsvarianten (z.B. Tests zur Eigenanwendung in verschiedenen Ländern für diverse Distributoren mit länderspezifischer Kennzeichnung) sowie Packungsvarianten des gleichen Produkts (z.B. 96-Well bzw. 128-Well Platten, Mehrfachabpackungen) vergeben.
Insbesondere hinsichtlich derselben wesentlichen Auslegungs- und Herstellungsmerkmale möchten wir für Medizinprodukte auf einige Aspekte hinweisen, welche zu unterschiedlichen Basis-UDI-DI führen:
Verbindliche Festlegung der deutschen Marktaufsichtsbehörden (AGMP) zum Umgang mit Konformitätserklärungen für Richtlinien-Produkte (gemäß 93/42/EWG bzw. 98/79/EG)
Zur Zeit besteht keine Notwendigkeit eine aktuelle Konformitätserklärung, wenn es keine Änderungen gibt, ergänzend zur ursprünglichen, vor dem Geltungsbeginn der MDR (26.05.2021) bzw. IVDR (26.05.2022) ausgestellten Konformitätserklärung, mit Bezug auf Art. 120 MDR bzw. Artikel 110 IVDR zu fordern.
Jedoch muss im Falle nicht signifikanter Änderungen, die eine Anpassung der Konformitätserklärung erfordern (z.B. Umfirmierung, Adressänderung) die bestehende Konformitätserklärung gemäß Richtlinie um ein Beiblatt bzw. Anhang ergänzt werden.
Dies wurde nun durch die deutschen Marktaufsichtsbehörden (AGMP) klargestellt und es wird besonders daraufhin gewiesen, dass es KEINE neue Konformitätserklärung nach MDD/ IVDD geben darf, sondern nur ein „Beiblatt“ (s.o.) zu der vor dem 2021-05-26 (MDD)/ 2022-05-26 (IVDD) ausgestellten Konformitätserklärung, welche die nicht signifikanten Änderungen (wie auch neue Produktvarianten) eindeutig nennt.
PSUR / Sicherheitsberichte für MDR-/ IVDR-zertifizierten Produkten
Erinnerung an die Notwendigkeit zur Einreichung von Sicherheitsberichten
Hersteller von Medizinprodukten der Risikoklassen IIa, IIb und III müssen gemäß MDR Art. 86 bzw. Hersteller von In-vitro-Diagnostika der Risikoklassen C und D müssen gemäß IVDR Art. 81 in einer bestimmten Frequenz Sicherheitsberichte erstellen. Für MDR-/ IVDR-zertifizierte Produkte sind diese Sicherheitsberichte bei der Benannten Stelle einzureichen und werden von ihr gemäß der Anforderungen geprüft.
Das MDCG 2022-21 enthält umfassende Erläuterungen zum Inhalt des Sicherheitsberichts für Medizinprodukte.
Für die MDR-/ IVDR-zertifizierten Produkte, für die mdc (unter Berücksichtigung der Erstellungsfristen) noch keine Sicherheitsberichte erhalten hat, wird mdc auf die Kunden zukommen und an die Notwendigkeit und Fristen erinnern.
Fristen für den Übergang der Zertifizierung von MDD zur MDR
Am 20. März 2023 wurde die Verordnung (EU) 2023/607 veröffentlicht, um den Übergang von der MDD (und AIMDD) zur MDR-Zertifizierung von auf dem Markt befindlichen Produkten zu erleichtern und die Verfügbarkeit von Medizinprodukten auf dem europäischen Markt zu gewährleisten. Hersteller von Produkten, die nach der Medizinprodukterichtlinie (MDD) zertifiziert sind (sog. “legacy devices” gemäß Artikel 120 der MDR) können nun von längeren Fristen für den Übergang von einer MDD-Zertifizierung zur MDR profitieren. Um von dieser Verlängerung profitieren zu können, sollten einige wesentliche Punkte genauer beachtet werden:
Verlängerung der Gültigkeit von MDR-Zertifikaten:
Alle Zertifikate, die am 20. März 2023 nicht abgelaufen waren, wurden per Definition bis zum 31. Dezember 2027 (für implantierbare legacy devices der Klassen III und IIb) bzw. bis zum 31. Dezember 2028 (für andere legacy devices der Klassen IIb und IIa sowie der Klassen Im und Is) verlängert. Zu beachten ist, dass eine zusätzliche Voraussetzung für das Inverkehrbringen von Produkten eingeführt wurde: Zusätzlich zur gültigen MDD-Bescheinigung muss der Hersteller für das einzelne Produkt (oder ein Nachfolgeprodukt) bis zum 26. Mai 2024 eine MDR-Zertifizierung beantragt haben. Die benannte Stelle kann auf dieser Basis ein Bestätigungsschreiben („confirmation letter“ im Sinne des Q&A-Dokuments der Europäischen Kommission) ausstellen, das die Gültigkeit der MDD-Bescheinigung formal bestätigt.
Es ist jedoch wichtig zu beachten, dass die Verlängerung an bestimmte Bedingungen geknüpft ist, um von der neuen Übergangsfrist zu profitieren. Die Hersteller müssen:
- bis 26. Mai 2024 ein Qualitätsmanagementsystem (QMS) gemäß MDR einrichten;
- bis 26. Mai 2024 einen förmlichen Antrag auf Konformitätsbewertung gemäß MDR stellen;
- bis 26. September 2024 eine von der Benannten Stelle und dem Hersteller schriftliche Vereinbarung für die Konformitätsbewertung nach der MDR unterzeichnet haben.
Außerdem gilt die Verlängerung der Übergangsregelung ausschließlich für MDR-Altgeräte, welche:
- die Richtlinie 93/42/EWG weiterhin einhalten;
- hinsichtlich Konstruktion oder Zweckbestimmung nicht wesentlich geändert wurden.
- keine unannehmbaren Risiken für die Gesundheit und Sicherheit von Patienten und Benutzern darstellen.
Mit der neuen Verordnung werden auch zusätzliche Anforderungen für Altprodukte mit abgelaufenen Zertifikaten eingeführt. Auch solche Produkte können von der verlängerten Übergangsfrist profitieren, wenn der Hersteller entweder vor Ablauf der MDD-Bescheinigung einen Vertrag mit einer benannten Stelle für die Konformitätsbewertung gemäß der MDR abgeschlossen hatte oder Ausnahmen gemäß Artikel 97 oder 59 MDR erhalten hat.
Diese Verlängerung der Übergangsfrist entspricht den Anforderungen der Medizinprodukteindustrie, die seit langem eine flexiblere Zertifizierungsfrist für MDD legacy devices gefordert hat. Dies ist zweifellos eine gute Nachricht für die Hersteller von Medizinprodukten. Es ist jedoch von entscheidender Bedeutung, rechtzeitig zu handeln und die erste Frist bis zum 26. Mai 2024 einzuhalten, um die Einhaltung der neuen Vorschriften zu gewährleisten.
In diesem Zusammenhang sei auch auf die Überarbeitung der MDCG 2020-3 (Rev. 1) zur Definition von wesentlichen Änderungen hingewiesen. Das Dokument klärt die Bedingungen, unter denen eine Änderung als “nicht signifikant” eingestuft werden kann und somit im Rahmen einer bestehenden MDD-Bescheinigung umgesetzt werden kann. Dies ist besonders wichtig für die Einführung von Produkten in den nächsten vier bis fünf Jahren, um die Sicherheit und Verfügbarkeit in der Übergangszeit zu gewährleisten.
Zusätzliche Übergangsfristen für Sonderanfertigungen:
Implantierbare Sonderanfertigungen der Klasse III müssen bis zum 26. Mai 2026 über eine Zertifizierung des Qualitätsmanagementsystems nach Anhang IX, Kapitel I&III MDR oder Anhang XI, Teil A MDR verfügen, um diese Produkte weiterhin auf den Markt bringen zu können.
Abschaffung der “Abverkaufsfrist”
Mit der neuen Verordnung wird die bisher in der MDR festgelegte “Abverkaufsfrist”, Artikel 120 Absatz 4 abgeschafft.
Zusätzliche Übergangsfristen für Medizinprodukte nach Anhang XVI:
Auch die Fristen für die Umsetzung der MDR für Produkte ohne medizinische Zweckbestimmung (Anhang XVI, MDR) wurden verlängert. Mit der Durchführungsverordnung (EU) 2022/2346 der Kommission wurde der rechtliche Rahmen, d.h. die gemeinsamen Spezifikationen, für die Zertifizierung von Anhang XVI-Produkten allgemein festgelegt. Die ursprüngliche Fassung wurde auch geändert, um die Übergangsfristen an die Definitionen der Verordnung (EU) 2023/607 anzupassen – diese wurde am 20. Juni 2023 als Durchführungsverordnung (EU) 2023/1194 der Kommission veröffentlicht.
Es wurde klargestellt, dass auch in diesem Fall für MDD-zertifizierte Medizinprodukte die Fristen des Art. 120 MDR (siehe oben) gelten. In allen anderen Fällen (nicht MDD-zertifizierte Produkte) werden auch die Fristen für den Übergang der Geräte zur MDR-Zertifizierung verlängert.
Voraussetzungen:
Das Produkt wurde vor dem 22. Juni 2023 rechtmäßig in der Union in Verkehr gebracht,
ist in kontinuierlicher Übereinstimmung mit den geltenden Anforderungen und
wurde nicht wesentlich geändert.
Anwendbare Fristen für Anhang-XVI-Produkte, bei denen der Hersteller eine klinische Prüfung (KP) durchführen will oder bereits durchführt:
22. Juni 2024: Bestätigung durch die zuständige Behörde gemäß Art. 70 (1) oder (3) MDR, dass der Antrag für die KP vollständig ist;
23. Dezember 2024: der Sponsor muss die KP beginnen;
1. Januar 2028: eine schriftliche Vereinbarung über die Konformitätsbewertung mit der Benannten Stelle muss unterzeichnet sein;
31. Dezember 2029: Ende der Übergangsfrist.
Anwendbare Fristen für Anhang-XVI-Produkte, bei denen der Hersteller keine klinische Prüfung durchführt:
1. Januar 2027: eine schriftliche Vereinbarung über die Konformitätsbewertung mit der Benannten Stelle muss unterzeichnet sein;
31. Dezember 2028: Ende der Übergangsfrist
Verlängerung der Übergangsfristen der MDR
Am 20. Mar. 2023 wurde von der Europäischen Kommission die Verordnung (EU) 2023/607 zur Verlängerung der Übergangsfristen für Produkte, welche nach der Verordnung (EU) 2017/745 (MDR) eine Benannte Stelle für die Konformitätsbewertung benötigen, veröffentlicht.
UPDATE: Im Juli 2023 hat die EU-Kommission zusätzlich ein aktualisiertes FAQ-Dokument zum Thema der Verlängerung der Übergangsfristen zur Verfügung gestellt, welches Sie hier finden können:
Die neue Verordnung regelt die Gültigkeit von AIMDD- und MDD-Zertifikaten neu und definiert Anforderungen für das erstmalige Inverkehrbringen von „Altprodukten“ (sog. „Legacy Devices“ im Sinn des MDCG 2021-25), auch wenn die Gültigkeit der zugrundeliegenden Zertifikate nach Richtlinie (MDD oder AIMDD) formal abgelaufen ist.
Überblick der Änderungen:
- Verlängerung der Übergangsfristen für das erstmalige Inverkehrbringen von Produkten auf Grundlage von Zertifikaten nach MDD oder AIMDD, für alle Produkte, für die vor Ablauf des MDD-Zertifikats ein Antrag auf Zertifizierung (siehe unten) nach MDR gestellt wurde bis 31. Dez. 2027 (Klasse III und implantierbare Produkte Klasse IIb [mit Ausnahmen]) bzw. bis 31. Dez. 2028 (alle anderen Produkte)
- Verlängerung der Übergangsfrist für Produkte der Klasse I (MDD), welche unter MDR höher klassifiziert werden bis 31. Dez. 2028
- Verlängerung der Übergangsfrist für Sonderanfertigungen der Klasse III bis 31. Dez. 2027
Für diese Produkte (ausgenommen Sonderanfertigungen) muss der Hersteller spätestens ab 26. Mai 2024 ein QM-System gemäß MDR etabliert haben. Die zweite, wesentliche Voraussetzung, um die Verlängerungen in Anspruch nehmen zu können, ist ein schriftlicher Vertrag zwischen dem Hersteller und der Benannten Stelle auf Zertifizierung nach MDR („Zertifizierungsvertrag“). Dieser Vertrag muss spätestens vor Ablauf des MDD-Zertifikates beantragt werden und alle Produkte umfassen, für welche die Verlängerung der Übergangsfristen in Anspruch genommen werden soll. Grundlage des Vertrags ist das Vorliegen der Technischen Dokumentationen gemäß den Anforderungen der MDR.
Um einen Antrags auf Zertifizierung – entweder erstmalige Zertifizierung nach MDR oder Erweiterung der MDR-Zertifizierung um neue Produktgruppen – stellen zu können, benötigen wir einen Fragebogen (erstmalige Zertifizierung) bzw. eine ergänzte Liste der Medizinprodukte (eine Änderungsmitteilung alleine ist nicht ausreichend). Auf dieser Basis werden wir Ihnen ein Angebot für die Zertifizierung erstellen. Dieses Angebot ist die Basis für Ihren Antrag auf Zertifizierung und in weiterer Folge für das finale Schließen eines Zeritifizierungsvertrages. Die Voraussetzungen zur Vertragsgestaltung werden wir dermaßen implementieren, dass gemeinsam mit dem Antrag auf Zertifizierung neben der Dokumentation des QM-Systems mindestens eine vollständige technische Dokumentation zur Prüfung eingereicht werden muss. Die Technischen Dokumentationen gemäß MDR-Anforderung für die weiteren vom Zertifizierungsvertrag erfassten Produkte müssen ebenfalls zur Antragstellung zur Verfügung stehen, können aber gemäß einem vereinbarten Plan zu einem späteren Zeitpunkt zur Prüfung übermittelt werden.
Um den von der EU-Kommission vorgeschlagenen Bestätigungsbrief („confirmation lettter“) ausstellen zu können, muss zusätzlich ein Vertrag für die Überwachung der (formal ausgelaufenen) MDD-Zertifikate geschlossen werden. Nur auf dieser Grundlage kann eine Bestätigung ausgestellt werden, dass Produkte weiterhin mit dem CE-Kennzeichen und der Kennnummer der mdc 0483 in Verkehr gebracht werden dürfen. Wir bereiten die entsprechenden Antrags-Verfahren dafür vor und werden diese so rasch wie möglich zur Verfügung stellen.
Alternativ kann der Hersteller (oder sein europäischer Bevollmächtigter) eine Ausnahmegenehmigung nach Art. 97 MDR bei der zuständigen Marktaufsichtsbehörde beantragen, diese Option ist unabhängig vom Status der MDD-Zertifikate.
Eine weitere wesentliche Neuerung ist ebenso der Wegfall der Abverkaufsfrist für unter AIMDD, MDD und IVDD in Verkehr gebrachte Medizinprodukte und In-vitro-Diagnostika.
Den Text der Verordnung finden Sie auch auf
https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:32023R0607
Produkte ohne medizinischer Zweckbestimmung – Verfahren nach Anh. XVI
Mit 1. Dez. 2022 hat die Europäische Kommission die Festlegungen gemeinsamer Spezifikationen für die in Anhang XVI der MDR genannten Produkte ohne medizinische Zweckbestimmung (Verordnung (EU) 2022/2346 veröffentlicht. Die Durchführungsverordnung enthält insbesondere Vorgaben für das Konformitätsbewertungsverfahren und die dabei zu berücksichtigenden Anforderungen – sogenannte gemeinsame Spezifikationen.
Insbesondere für all jene Produkte, welche bisher nicht dem Medizinprodukterecht unterlagen, aber die Anforderungen der MDR erfüllen müssen, werden spezifische Voraussetzungen und Übergangsfristen festgelegt. Grundvoraussetzung ist, dass das Produkt vor dem 22. Juni 2023 bereits rechtmäßig in der Union in Verkehr gebracht wurde und weiterhin die gesetzlichen Anforderungen erfüllt. Ebenso dürfen die Auslegung und Zweckbestimmung des Produkts keine wesentlichen Änderungen (im Sinn des Art. 120 (3) MDR sowie MDCG 2020-3) erfahren. Diese Produkte dürfen gemäß Art. 2 (2) VO (EU) 2022/2346 ab dem 22. September 2023 bis zum 22. Juni 2025 dann in Verkehr gebracht oder in Betrieb genommen werden, wenn eine schriftliche Vereinbarung über die Durchführung der Konformitätsbewertung zwischen der Benannten Stelle und dem Hersteller unterzeichnet wurde.
Besondere Fristen wurden darüber hinaus für Produkte festgelegt, für welche im Rahmen der erstmaligen Konformitätsbewertung als Medizinprodukt eine klinische Prüfung durchgeführt wird.
Des Weiteren dürfen wir auf die aktuelle europäische Diskussion betreffend der Übergangsfristen für bereits MDD-zertifizierte Produkte verweisen, welche auch Produkte betreffen kann, welche unter Anh. XVI MDR geregelt sind.
Wir werden unsere Bestandskunden rechtzeitig zum Wahren dieser Fristen über die Möglichkeiten und Verfahren bei mdc informieren.
MDR – EU-Kommission fordert Hersteller zur Antragstellung auf
Mit dem neuen Positionspapier MDCG 2022-11, welches von der Europäischen Kommission veröffentlicht wurde, werden Hersteller aufgefordert, Ihre Anträge für eine Zertifizierung unter der MDR frühzeitig und vollständig zu stellen.
Damit trägt die MDCG der Tatsache Rechnung, dass gemäß einer Auskunft von TEAM-NB, dem europäischen Verband der Benannten Stellen für Medizinprodukte, die meisten Mitglieder mitteilten, dass mehr als die Hälfte der Anträge unvollständig sind, d. h. dass sich die QM-Dokumentation oder Technische Dokumentation in einem nicht begutachtungsfähigen Zustand befindet.
Es wird darauf hingewiesen, dass die Benannten Stellen nicht in der Lage sein dürften, alle Dokumentationen bis zum Mai 2024 zu prüfen. Ferner wird klargestellt, dass eventuelle Ausnahmegenehmigungen durch die nationalen Behörden nur dann erfolgen können, wenn die betreffenden Produkte im Sinne der öffentlichen Gesundheit, der Patientensicherheit oder Patientengesundheit tatsächlich benötigt werden.
Der vor diesem Hintergrund gemachte Aufruf der schnellstmöglichen Einreichung von vollständigen und konformen Dokumentationen wird seitens mdc aufgrund der bisherigen MDR-Erfahrung deutlich unterstützt und es wird ergänzend darauf hingewiesen, dass eine vollständige Einreichung noch 2022 bei mdc erfolgen sollte, um die Chance auf eine Zertifizierung bis Mai 2024 zu wahren.
25.01.2022: EU ändert Übergangsbestimmungen der IVDR
Mit der Verordnung (EU) 2022/112 hat die EU die Übergangsbestimmungen für bestimmte In-vitro-Diagnostika geändert und den Geltungsbeginn der IVDR für „hausinterne Produkte“ bis zum 26. Mai 2028 aufgeschoben. Obgleich der Anwendungsbeginn am 26.05.2022 bestehen bleibt, erlaubt der neue Rechtsrahmen für viele In-vitro-Diagnostika, die sich bereits im Verkehr befinden, längere Übergangsfristen:
- Bescheinigungen, die von Benannten Stellen gemäß Richtlinie 98/79/EG ausgestellt wurden, verlieren erst spätestens am 27. Mai 2025 ihre Gültigkeit. Dies bedeutet, dass Produkte, welche unter der Richtlinie 98/79/EG durch eine Benannte Stelle zertifiziert wurden, bis zum 26. Mai 2025 in Verkehr gebracht oder in Betrieb genommen werden können.
- Altprodukte, welche vor dem 26.05.2022 über eine gültige Konformitätserklärung gemäß Richtlinie 98/79/EG verfügten und ohne das Mitwirken einer Benannten Stelle auf dem Markt bereitgestellt wurden, dürfen in Abhängigkeit von der Risikoklasse bis zu folgenden Zeitpunkten in Verkehr gebracht werden:
- 26. Mai 2025 für Produkte der Klasse D;
- 26. Mai 2026 für Produkte der Klasse C;
- 26. Mai 2027 für Produkte der Klasse B;
- 26. Mai 2027 für Produkte der Klasse A, die in sterilem Zustand in Verkehr gebracht werden.
Zusätzlich wurden Fristen für die Bereitstellung auf dem Markt und die Inbetriebnahme festgelegt.
Für das Änderungsmanagement der Produkte, die gemäß Richtlinie 98/79/EG in Verkehr gebracht oder in Betrieb genommen werden, erwarten wir analog zu MDCG 2020-3 (Guidance on significant changes regarding the transitional provision under Article 120 of the MDR with regard to devices covered by certificates according to MDD or AIMDD) ein ähnliches Guidance Dokument.
Die Anforderungen der IVDR an die Überwachung nach dem Inverkehrbringen, die Marktüberwachung, die Vigilanz sowie die Registrierung von Wirtschaftsakteuren und von Produkten gelten jedoch für alle IVD ab dem 26.05.2022.
25 Jahre mdc medical device certification GmbH
Die Gründung der Zertifizierungsgesellschaft mdc medical device certification GmbH wurde heute vor 25 Jahren, am 10. Dezember 1996 zur Eintragung beim Handelsregister Memmingen angemeldet.
Bereits 1994 war „mdc medical device certification“ die Bezeichnung eines Geschäftsbereichs der Dr. Müller-Lierheim GmbH, welcher mit der 1996 vorgenommenen Unternehmensgründung in die rechtliche Eigenständigkeit überführt wurde.
Seit der Fusion mit der Zertifizierungsstelle Medizinprodukte von ZDH-ZERT e.V. im Jahre 2000 ist der Sitz der mdc in Stuttgart. Weitere Bürostandorte befinden sich in Berlin, Tuttlingen und Wien. Ferner wird in wenigen Wochen in Haifa (Israel) die erste außereuropäische Niederlassung bezogen. In den vergangenen 25 Jahren ist die Anzahl der Angestellten von drei auf über 100 gestiegen. Zusätzlich sind ungefähr 70 freiberuflich tätige Auditoren, Fachexperten und Begeher tätig. Das Tochterunternehmen des ZDH-ZERT Verein für Qualität im Handwerk und in der gewerblichen Wirtschaft e.V. gehört nicht nur zu den führenden Benannten Stellen und Präqualifizierungsstellen sondern ist auch Marktführer bei der Zertifizierung von QM-Systemen in Betrieben der Gesundheitshandwerke und Hilfsmittelversorger.
Die mdc hat ihre Aktivitäten stets auf das Gebiet der Medizinprodukte und verwandte Bereiche fokussiert. Derzeit deckt das Spektrum die Tätigkeit als Benannte Stelle unter der Verordnung (EU) 2017/745 für Medizinprodukte (MDR) und unter der Richtlinie 98/79/EG für In-vitro Diagnostika (IVDD), als akkreditierte Zertifizierungsstelle für QM-Systeme gemäß ISO 13485 und ISO 9001 sowie als akkreditierte Präqualifizierungsstelle im Gebiet der Hilfsmittelversorgung ab. Abgerundet wird das Angebot durch Anerkennungen in der Ukraine und in Taiwan, durch Audits unter dem Medical Device Single Audit Program (MDSAP) im Rahmen eines Kooperationsverfahrens sowie die Veranstaltung öffentlicher Präsenz- und Onlineseminare zu Themen aus dem Bereich Qualitätsmanagement und Regulatory Affairs. Unter der Verordnung (EU) 2017/746 (IVDR) befindet sich mdc in einem fortgeschrittenen Stadium des Benennungsverfahrens.
Anlässlich des 25-jährigen Firmenjubiläums durfte der Geschäftsführer Harald Rentschler, der diese Position bei der Gründung antrat, von der Geschäftsführerin der IHK Stuttgart, Frau Dr. Susanne Herre eine Ehrenurkunde der IHK entgegen nehmen. Er dankt allen, die das Unternehmen auf seinem bisherigen erfolgreichen Weg begleitet haben.
Legacy Devices
Sehr geehrte Damen und Herren!
Mit 2021-05-26 wurde die Richtlinie 93/42/EWG über Medizinprodukte (im weiteren MDD) zurückgezogen und damit ungültig. Damit einhergehend wurde gemäß Artikel 120 (1) der Verordnung (EU) 2017/745 über Medizinprodukte (im weiteren MDR) auch die Benennung aller Benannten Stellen nach MDD und auch unsere Benennung aufgehoben.
Das bedeutet erstens, dass wir als Ihre Benannte Stelle gemäß MDD keine neuen Zertifikate oder Erweiterungen von Zertifikaten mehr ausstellen können. Die Gültigkeit der bisher ausgestellten Zertifikate bleibt weiterhin bestehen, wenn diese gemäß Artikel 120 (3) MDR, letzter Satz, einer angemessenen Überwachung durch die Benannte Stelle unterliegen.
Zweitens bedeutet diese Situation auch, dass ein wesentlicher Bestandteil des Zertifizierungsvertrages weggefallen ist, da mdc keine Benannte Stelle nach MDD mehr ist. (Die Benennung als Benannte Stelle nach MDR sowie IVDD ist davon unberührt und bleibt weiterhin bestehen!) Daher werden wir in den nächsten Tagen mit einer Zusatzvereinbarung zu dem existierenden Zertifizierungsvertrag auf Sie zukommen, um die nach Artikel 120 (3) MDR geforderte Überwachung auf einer vertraglich fundierten Basis zu ermöglichen. Dies ist eine wesentliche Voraussetzung für die weitere Gültigkeit der ausgestellten Zertifikate nach MDD.
Des Weiteren möchten wir Sie mit dieser Aussendung über neue MDCG-Guidance-Dokumente informieren. Die MDCG-Guidance-Dokumente stellen eine Hilfestellungen zur harmonisierten Interpretation des Gesetzestextes der MDR durch die europäische Kommission und zuständigen Behörden dar. MDCG-Guidance-Dokumente sollen bei der Umsetzung eines QM-Systems sowie bei der Erstellung und Pflege der Technischen Dokumentation berücksichtigt werden.
Mit 2021-10-21 wurde das lange erwartete MDCG 2021-25[1] über den Umgang mit „Legacy Produkten“ veröffentlicht. Unter „Legacy Produkten“ werden dabei solche Produkte verstanden, welche eine Zertifizierung nach MDD besitzen und in der, gemäß Artikel 120 (3) MDR genannten Übergangsfrist, unter den dort genannten Voraussetzungen noch bis zum Ende der Laufzeit des jeweiligen Zertifikats bzw. der Übergangsfrist unter MDD-Anforderungen in Verkehr gebracht werden dürfen. Solche Legacy Produkte können weiterhin in Verkehr gebracht werden, sofern die Sicherheit und Leistungsfähigkeit unter den Vorgaben der MDD belegt ist. Wir haben dazu schon mehrere News-Artikel auf unserer Homepage veröffentlicht, auf die wir hinweisen dürfen z.B.
- 26.05.2021: Geltungsbeginn der neuen Medical Device Regulation
- Änderungen bei MDD-Verfahren auf Grund des Geltungsbeginns der MDR
Die Vorgaben des Artikels 120 (3) MDR enthielten jedoch in einigen Punkten einen Interpretationsspielraum, welcher durch das MDCG 2021-25 klargestellt wurde.
1. Verantwortliche Person nach Art. 15
Erste und wichtigste Klarstellung ist, dass für diese Legacy-Produkte keine „für das Einhalten der Regulierungsvorschriften verantwortliche Person“ (PRRC gemäß Art. 15 MDR bzw. IVDR) benannt werden muss.
Diese Regelung des Artikel 15 MDR und IVDR löst in Deutschland und Österreich die Notwendigkeit der Bestellung eines Sicherheitsbeauftragten ab. Die bisherige Anforderung zur Benennung eines Sicherheitsbeauftragten (z.B. nach § 31 MPG bzw. § 78 österr. MPG 1996) besteht seit 2021-05-26 nur noch für Hersteller von In-vitro-Diagnostika; das MPDG[2], wie auch das österreichische MPG 2021[3] enthalten diese Forderung nicht mehr. Wir empfehlen dennoch weiterhin, dass auch Hersteller, welche ausschließlich „Legacy-Produkte“ in Verkehr bringen, die Prozesse anpassen. Insbesondere sollten die Verantwortlichkeiten für das Erfüllen der Meldepflichten eindeutig einer „Verantwortlichen Person“ zugewiesen werden. Da diese Person jedoch rechtlich noch nicht verpflichtend ist, sehen wir es ebenso noch nicht verpflichtend, dass die in der MDR genannten Qualifikationsanforderungen vollinhaltlich eingehalten werden.
Handelt es sich bei dem Unternehmen um einen Hersteller von Produkten der Klasse I, welche seit 2021-05-26 vollinhaltlich der MDR unterliegen (also keine Höherklassifizierung durch die MDR), so sind die Vorgaben des Artikels 15 MDR in jedem Fall vollinhaltlich anwendbar.
2. Regelmäßig aktualisierter Sicherheitsbericht – PSUR nach Art. 86
Eine weitere Klarstellung betrifft die Vorgaben für die Überwachung nach dem Inverkehrbringen – Post Market Surveillance (PMS). Die Anforderungen der Artikel 82 bis 86 MDR sowie des Anhang III müssen seit 2021-05-26 durch jeden Hersteller von Medizinprodukten erfüllt werden. Das bedeutet, dass auch für Legacy-Produkte ein PMS-Plan erstellt und als Bestandteil der Technischen Dokumentation dokumentiert werden muss. Die detaillierten Inhalte sind im Anhang III der MDR vorgeben.
Die zweite Anforderung ist, dass gemäß den Vorgaben dieses Planes für alle Medizinprodukte der Klassen IIa, IIb und III gemäß Artikel 86 MDR ein „regelmäßig aktualisierter Bericht über die Sicherheit“ (PSUR) erstellt und aktualisiert werden muss. Durch das MDCG-Guidance-Dokument wurde eindeutig klargestellt, dass dieser Bericht auch von Herstellern von „Legacy-Produkten“ erwartet wird.
Dieser PSUR muss den Marktaufsichtsbehörden auf Anfrage zur Verfügung gestellt werden. Der Benannten Stelle muss der PSUR für „legacy-Produkte“ im Rahmen von Überwachungsaudits zugänglich gemacht werden.
Bei mdc werden wir die fristgerechte Umsetzung dieser Vorgaben im Rahmen des Audits detailliert prüfen. Das bedeutet, dass für „Legacy-Produkte“ seit 2021-05-26 ein PMS-Plan vorliegen muss, die Inhalte des Plans müssen die inhaltlichen Anforderungen des Anhang III der MDR erfüllen. Ebenso muss der Plan das Datum der ersten Erstellung bzw. das Intervall für die Aktualisierung des PSUR angeben. Die Fristen zur erstmaligen Erstellung und weiteren Aktualisierung dürfen dabei ein Jahr für Medizinprodukte der Klassen IIb und III sowie zwei Jahre für Medizinprodukte der Klasse IIa nicht überschreiten.
Ab dem im Plan festgelegten Datum muss auch der PSUR vorliegen und wird im Rahmen des Audits inhaltlich (stichprobenartig) geprüft werden. Dabei sind neben der Zusammenschau über meldepflichtige / schwerwiegende Vorkommnisse weitere Informationen dazulegen. Dies beinhaltet beispielsweise
- eine Übersicht der nicht-schwerwiegenden Vorkommnisse,
- ein Trending nicht schwerwiegender Vorkommnisse oder erwarteter unerwünschter Nebenwirkungen,
- die Schlussfolgerungen aus der Nutzen-Risiko-Abwägung,
- die wichtigsten Ergebnisse des PMCF-Bewertungsberichts, sofern verfügbar, und
- die Gesamtabsatzmenge des Produkts und eine Schätzung der Anzahl der Anwendungen / Anwendungshäufigkeit.
3. Umgang mit Änderungen an Legacy Produkten
An dieser Stelle möchten wir nochmals an die Bedingungen erinnern, wie für „Legacy-Produkte“ das MDD-Zertifikat weiterhin aufrechterhalten werden kann. Neben der eingangs erwähnten, fortlaufenden Überwachung durch die Benannte Stelle im Rahmen von Audits sowie Stichprobenprüfungen der Technischen Dokumentation dürfen die Produkte keine wesentliche Änderung („significant change“), wie in Artikel 120 (3) definiert und in MDCG 2020-3[4] näher ausgeführt, erfahren. Insbesondere MDCG 2020-3 enthält eine detaillierte Beschreibung einschließlich Beispielen und einem Entscheidungsbaum zur Bewertung einer Änderung. Das Umsetzen einer „significant change“ führt dazu, dass das ausgestellte MDD-Zertifikat seine Gültigkeit verliert. Solch eine Änderung darf nur im Rahmen einer Zertifizierung nach MDR umgesetzt werden.
Davon unabhängig besteht grundsätzlich die Verpflichtung zur Meldung von Änderungen am Produkt, am QM-System, an der Produktpalette usw., welche im Guidance Dokument NBOG 2014-3[5] beschrieben sind. Solche „substancial changes“ werden weiterhin durch die Benannte Stelle bewertet, bevor diese implementiert werden können. Änderungen, welche zwar als „substancial change“ meldepflichtig sind, aber nicht die Definition eines „significant changes“ erfüllen, können nach einer Bewertung durch die Benannte Stelle umgesetzt werden. Bitte kontaktieren Sie im Zweifelsfall das Projektteam.
Im Rahmen des Audits vor Ort wird insbesondere geprüft, ob das Unternehmen Prozesse implementiert hat, um geplante Änderungen zu klassifizieren und, wenn erforderlich, diese der Benannten Stelle anzuzeigen. Des Weiteren sollen die geplanten und umgesetzten Änderungen hinsichtlich ihrer Klassifizierung (significant und/oder substancial) bewertet werden. Der Auditor/die Auditorin müssen die Validität der Einstufung durch den Hersteller bestätigen.
4. Umstellung auf die MDR
Wir möchten an dieser Stelle nochmals daran erinnern, dass die Übergangsfristen für Zertifikate nach der MDD spätestens mit 2024-05-26 auslaufen. Bis zu diesem Zeitpunkt muss für alle Produkte der Klassen Im, Ir, Is, IIa, IIb und III mindestens ein Zertifikat nach MDR vorliegen, wenn diese weiterhin in Europa in Verkehr gebracht werden sollen. Für Medizinprodukte der Klasse III müssen beide Zertifikate (Bewertung der technischen Dokumentation und QM-System) nach demselben Regelwerk (MDD oder MDR) gültig sein.
Bei den Zertifizierungsverfahren nach MDR handelt es sich um wirkliche Neuzertifizierungen. Die Verfahren sind sowohl formal als auch inhaltlich mit wesentlich höheren Aufwänden verbunden und benötigen daher signifikant mehr Zeit als unter MDD. Die Erfahrungen aus den ersten 1,5 Jahren haben auch gezeigt, dass die Anforderungen an die Neugestaltung der Technischen Dokumentation immer wieder unterschätzt wurden und daher TD-Prüfberichte mit einer sehr hohen Zahl an Abweichungen keine Seltenheit sind. Um die Zertifizierungsverfahren nach MDR bis zum Ende der Gültigkeit der MDD-Zertifikate rechtzeitig abschließen zu können möchten wir Sie daher ersuchen, das geplante Vorgehen mit Ihrem mdc-Projektteam frühzeitig abzustimmen – nur mit einer Einreichung der QM-Dokumentation und aller Technischen Dokumentation im Jahr 2022 kann die Möglichkeit einer Zertifizierung unter der MDR bis zum Mai 2024 gewahrt werden. Das Erfordernis einer frühzeitigen Einreichung müssen wir mit der Tatsache auch dahingehend unterstreichen, dass wir derzeit schon einzelnen Bestandskunden aus Ressourcengründen keine Zertifizierung nach der MDR anbieten können.
Für Rückfragen stehen wir selbstverständlich gerne zur Verfügung
Daniel Kraushaar Meinrad Guggenbichler
Bereichsleitung Medizinprodukte Leitung Benannte Stelle Medizinprodukte
[1] MDCG 2021-25: Application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021: https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_2021_25_en.pdf
[2] MPDG: Gesetz zur Durchführung unionsrechtlicher Vorschriften betreffend Medizinprodukte: https://www.gesetze-im-internet.de/mpdg/
[3] MPG 2021: Medizinproduktegesetz 2021: https://www.ris.bka.gv.at/GeltendeFassung.wxe?Abfrage=Bundesnormen&Gesetzesnummer=20011580
[4] MDCG 2020-3: Guidance on significant changes regarding the transitional provision under Article 120 of the MDR: https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_guidance_significant_changes_annexes_en.pdf
[5] NBOG 2014-3: Guidance for manufacturers and Notified Bodies on reporting of Design Changes and Changes of the Quality System: http://www.doks.nbog.eu/Doks/NBOG_BPG_2014_3.pdf