
CE-Kennzeichnung
Verordnung (EU) 2017/746 IVDR
IVDR (EU) 2017/746 Zertifizierung: Die Verordnung IVDR ist die gültige regulatorische Grundlage für In-Vitro-Diagnostika (IVD) auf dem europäischen Markt. Um den immer stärker steigenden Anforderungen der Gesellschaft an die Sicherheit und Zuverlässigkeit von IVD gerecht zu werden, trat 2017 die Verordnung (EU) 2017/746 über In-vitro-Diagnostika (IVDR) in Kraft und löste die frühere EU-Richtlinie 98/79/EG (IVDD) ab. Am 25. Mai 2022 endete die Übergangsfrist, jedoch wurden verlängerte Übergangsfristen für Produkte der Klasse A steril sowie B, C und D festgelegt. Als eine der wenigen Benannten Stellen in Europa ist mdc Ihr kompetenter Partner im Rahmen der CE-Zertifizierung nach den neuen Regularien (EU) 2017/746.
Direkt zum Ansprechpartner
Noch Fragen zum Produkt? Hier geht’s direkt zum Ansprechpartner.
Leistungsspektrum für die CE-Zertifizierung von In-vitro-Diagnostika
Für Hersteller von In-vitro-Diagnostika führen wir die im Rahmen der CE-Kennzeichnung erforderlichen Zertifizierungen nach der Verordnung (EU) 2017/746 als Benannte Stelle durch. Konformitätsbewertungsverfahren mit Benannten Stellen sind für alle Hersteller von Produkten der Klasse A (steril) sowie der Klassen B, C und D verpflichtend. Unser Angebot umfasst auch die Zertifizierung von patientennahen Tests und Produkten zur Eigenanwendung durch Laien.
Schwerpunkte unserer Tätigkeit sind hierbei sowohl die Überprüfung des QM-Systems in den Betriebsstätten des Herstellers und seiner Unterauftragnehmer als auch die Überprüfung der Technischen Dokumentationen zu den jeweiligen Produkten entsprechend Anhang IX. Dazu stehen uns neben produkt- und technologieerfahrenden Auditoren qualifizierte Fachexperten mit langjähriger Expertise in Industrie, Forschungsinstituten, Laboratorien oder bei Benannten Stellen zur Verfügung. Für die verpflichtende Überprüfung hergestellter Produkte unterstützen wir den Hersteller in der Zusammenarbeit mit benannten EU-Referenzlaboratorien.
| Klassifizierung | Von mdc angebotene Bewertungsverfahren |
|---|---|
| Klasse A (steril) | Anhang IX, Kapitel I.
(die Beteiligung der Benannten Stelle beschränkt sich auf Sterilitätsaspekte) |
| Klasse B | Anhang IX, Kapitel I und II
dies umfasst auch die Bewertung der technischen Dokumentation von mindestens einem repräsentativen Produkt pro Produktkategorie gemäß Anhang IX Abschnitt 4 |
| Klasse B für Eigenanwendung oder patientennahe Tests | Hier erfolgt zusätzlich eine Bewertung der technischen Dokumentation jedes Produkts für die spezifischen Aspekte gemäß Abschnitt 5.1 |
| Klasse C | Anhang IX, Kapitel I und II dies umfasst auch die Bewertung der technischen Dokumentation (von mindestens einem repräsentativen Produkt pro generischer Produktgruppe) gemäß Anhang IX Abschnitt 4 |
| Klasse C für Eigenanwendung oder patientennahe Tests | Hier erfolgt zusätzlich eine Bewertung der technischen Dokumentation jedes Produkts für die spezifischen Aspekte gemäß Abschnitt 5.1 |
| Klasse D | Anhang IX, Kapitel I und II
dies umfasst auch die Bewertung der technischen Dokumentation für jedes Produkt gemäß Anhang IX Abschnitt 4 (inkl. Überprüfung durch ein EU-Referenzlabor) |
| Klasse D für Eigenanwendung oder patientennahe Tests | Anhang IX, Kapitel I und II |
Unsere Kooperationspartner in der Ukraine erkennen Zertifizierungen der mdc für die nationale Produktzulassung an.
Leistungen für Drittänder
Produktzulassung in der Ukraine
An Zertifizierungen Interessierten bieten wir die Möglichkeit zu einem ersten Informationsaustausch.
KONTAKTIEREN SIE UNSER IVDR-TEAM
Ablauf eines Zertifizierungsverfahrens
Hier sehen Sie den beispielhaften Ablauf der Bearbeitung eines mdc Zertifizierungsverfahrens im Bereich In-Vitro-Diagnostika im Rahmen der CE-Kennzeichnung erforderlichen Zertifizierungen nach der Verordnung (EU) 2017/746
IVDR Dokumente
Übersicht von mdc-Dokumenten zur IVDR, die von Interesse sein könnten
01
Vorbereitung
- Anfrage
- Vorprüfung und Angebot
- Vertrag
- Antrag
02
Begutachtung
- Prüfung der Technischen Dokumentation (einschl. SSP)
- Besondere Verfahren (z.B. für innovative Produkte)
- Prüfung der QM-Dokumente
- Audit vor Ort (2-stufig)
- Begutachtungsbericht
03
Zertifizierung
- Bewertung aller Tätigkeiten und Berichte
- Entscheidung
- Zertifikatsausstellung
04
Überwachung
- Prüfung der Technischen Dokumentation (Stichproben)
- Sicherheitsberichte (PSUR)
- Überwachungsaudits
- Überprüfung der hergestellten Produkte (nur Klasse D)
- Begutachtungsberichte
- Bearbeitung von Änderungsmitteilungen
- Unangekündigte Audits
05
Re-Zertifizierung
- Vorbereitung
- Begutachtung
- Zertifizierung
Zusatzleistungen die von Interesse sein könnten
EN ISO 13485
Hier erhalten Sie weitere Informationen zum Produkt EN ISO 13485
Drittländer
2017/745 (MDR)
Hier erhalten Sie weitere Informationen zum Produkt 2017/745 (MDR)
Häufige Fehler bei der Einreichung von Technischen Dokumentationen für In-vitro-Diagnostika
Die nachfolgenden Punkte fassen häufige Mängel zusammen, welche mdc medical device certification GmbH bei der Einreichung von Technischen Dokumentationen für IVDs in den letzten zwei Jahren beobachtet haben. Diese Beispiele sollen mögliche Fallstricke verdeutlichen und stellen keine formale Empfehlung dar.
Fehler bei der Datenvollständigkeit und der Dateiübermittlung
- Dateien werden in falschen Formaten eingereicht, z.B. als *.docx oder in ZIP-Dateien mit unnötigen Unterordnern (Vgl. dazu die „Vorgaben für Einreichungen in digitaler Form“, abzurufen über das Service-Portal)
- Vorgaben zur maximalen Dateinamenslänge werden nicht eingehalten.
- Erforderliche Inhalte aus den Anhängen II und III fehlen oder sind unvollständig.
- Als nicht anwendbar markierte Punkte werden nicht oder nicht ausreichend begründet.
- Nachweise zur Erfüllung der Anforderungen werden nicht klar zugeordnet oder nicht eindeutig gekennzeichnet (z.B. UDI, Lieferanten, CoA).
- Produktmuster für Laienanwendungen und patientennahe Tests fehlen.
Allgemeine inhaltliche Mängel
- Die Gebrauchsanweisung wird häufig fälschlicherweise als Nachweis für die Produktbeschreibung, Probennahme oder Probenbearbeitung eingereicht.
- Unzureichende Angaben in der Produktbeschreibung, insbesondere zu:
• Produktabbildungen (Fotos, Zeichnungen),
• Bestandteilen und Zubehör (gemäß IVDR),
• Kombinationsgebrauch (Probenvorbereitung, Methoden, Geräte, Software),
• Markteinführungsart (Kit, Verkaufs- und Versandverpackung),
• den Ländern, in denen das Produkt vertrieben werden soll,
• der Klarheit von Produktvarianten in verschiedenen Dokumenten. - Die UDI ist unzureichend oder nicht für jedes Produkt klar definiert.
- Der Verwendungszweck ist nicht mit den Vorgaben der IVDR, Anhang I 20.4.1c) konform oder wird in verschiedenen Dokumenten unterschiedlich dargestellt.
- Die Klassifizierung des Produkts als IVD, sowie die zugehörige Regel zur Risikoklasse werden nicht ausreichend begründet.
- Rohmaterialien sind nicht korrekt zugeordnet, Spezifikationen und Verpackungsmaterialien fehlen oder sind unvollständig.
- Automatisierte Prozesse (Instrumente, Software) werden nicht ausreichend spezifiziert (z.B. keine Angabe von Versionsnummern der verwendeten Geräte, Anwendungen).
- Frühere Produktgenerationen oder Leistungsdaten von bereits auf dem Markt befindlichen Produkten werden nicht nachvollziehbar beschrieben.
- Nicht alle Kennzeichnungen und Gebrauchsanweisungen werden eingereicht, und Restrisiken sind darin nicht ausreichend deklariert (z.B. als Warnung oder Kontraindikation).
- Der Entwicklungs- und Herstellungsprozess wird nicht klar beschrieben, insbesondere ausgelagerte Prozesse und Qualitätskontrollen.
- Anforderungen der GSPR (Anhang I, IVDR) werden nicht vollständig oder nachvollziehbar begründet, und Nachweise sind oft unzureichend gekennzeichnet.
- Das Risikomanagement weist Lücken auf, wie unklare Verantwortlichkeiten, fehlende klinische Expertise und unzureichende Risikobewertungen, insbesondere bei älteren Produkten.
- Stabilitätsprüfungen sind nicht ausreichend dokumentiert, und Prüfpläne oder Testmethoden fehlen.
- Die Verwendung von Materialien menschlichen, tierischen oder mikrobiellen Ursprungs werden nicht hinreichend dokumentiert, und Sicherheitsnachweise sind unvollständig.
- Erkenntnisse aus der Post-Market-Surveillance (PMS) werden nicht in die Dokumentation integriert, und die Auswahl ähnlicher Produkte oder Datenbanken wird nicht begründet.
Mängel in der Darstellung der Leistungsbewertung
Ein häufig auftretender Fehler bei der Einreichung der technischen Dokumentation ist das Fehlen eines aussagekräftigen und strukturierten Leistungsbewertungsplans, insbesondere für sogenannte „Legacy-Produkte“, die bereits auf dem Markt sind. Es wird oft nicht deutlich, wie der Hersteller plant, die Konformität des Produkts nachzuweisen. Wichtige Details zur Verfügbarkeit und Nutzung historischer Daten, Literaturquellen, Ergebnisse von Ringversuchen oder nicht veröffentlichten internen Daten fehlen. Wenn Daten äquivalenter Produkte verwendet werden, wird die Äquivalenz der Produkte nicht detailliert dargelegt.
Leistungsbewertungsplan
- Die Gründe für nicht anwendbare analytische und klinische Leistungsmerkmale werden nicht ausreichend oder nachvollziehbar erläutert.
- Akzeptanzkriterien für analytische und klinische Studien sind nicht definiert oder es fehlt ein Verweis auf entsprechende Vorgaben.
- Statistische Rationalen für die Anzahl der verwendeten Proben oder Wiederholungen sowie die Anzahl und Qualifikation der Anwender oder Studiengruppen sind unzureichend dokumentiert. Auch Hinweise auf die angewandten Standards (z.B. Anhang, Tabelle oder Spalte) fehlen.
- Die Kriterien für die Annehmbarkeit des Risiko-Nutzen-Verhältnisses sind nicht festgelegt.
Analytische Leistung
- Häufig basieren die Nachweise auf Ergebnissen eines „Legacy Devices“, ohne dass Unterschiede zwischen dem beantragten Produkt und dem Legacy-Produkt klar aufgezeigt oder die Übertragbarkeit der alten Daten auf das IVDR-Produkt nachvollziehbar erläutert wird.
- Eine allgemeine Beschreibung des Studiendesigns ist oft unvollständig: Informationen zu Probenmaterial, Akzeptanzkriterien, Stichprobengröße sowie die Auswahl und Durchführung der Methodik fehlen oder sind nicht ausreichend erläutert. Verweise auf interne SOPs oder externe Richtlinien (z.B. CLSI) sind allein nicht ausreichend.
- Definitionen für Ausreißer und Ausschlusskriterien sind nicht angegeben. Maßnahmen für Wiederholungsmessungen werden nicht spezifiziert.
- Begründungen für das verwendete Vergleichsprodukt sowie die Auswahl der zu testenden interferierenden Substanzen und Kreuzreaktionen fehlen oft.
- Der Kombinationsgebrauch, insbesondere bei der Nutzung verschiedener Geräte oder Methoden in Verbindung, wird nicht umfassend berücksichtigt.
Klinische Leistungsbewertung
- Die Qualifikation der Studienleiter für klinische Leistungsstudien wird nicht immer ausreichend dargelegt.
- Die Vergleichbarkeit zwischen dem Legacy Device und dem beantragten Produkt wird nicht nachvollziehbar präsentiert.
- Wie bei der analytischen Leistung fehlen oft Definitionen für Ausreißer und Ausschlusskriterien sowie die Spezifikation von Maßnahmen für Wiederholungsmessungen.
- Eine nachvollziehbare Begründung, warum klinische Daten nicht in Anwenderhand oder in einer klinischen Umgebung erhoben wurden, fehlt.
- Studienberichte werden nicht von einer befugten Person oder einem Arzt/einer Ärztin unterzeichnet. Außerdem fehlen häufig Informationen zu Studiendetails wie Studienort, Zeitraum und verwendeten Materialien.
- Die Ergebnisse werden oft nur als oberflächliche Zusammenfassung dargestellt, ohne dass der Zusammenhang zwischen den einzelnen Studienergebnissen und den dokumentierten Prüfberichten klar ersichtlich ist.
- Abweichende oder ausgeschlossene Einzelergebnisse werden nicht ausreichend diskutiert oder erklärt.
Daten aus der Literatur und anderen Quellen
- Protokolle für die Literaturrecherche sind oft nicht verfügbar oder unvollständig; es fehlen definierte Suchwörter, Schlüsselbegriffe sowie Inklusions- und Exklusionskriterien.
- Der Stand der Technik wird dadurch nicht ausreichend belegt oder ist unvollständig dokumentiert.
- Wenn die klinische Leistung des Produkts ausschließlich durch Literaturquellen nachgewiesen wird, fehlt eine aussagekräftige Zusammenfassung der Literaturergebnisse, die sowohl positive als auch negative Ergebnisse berücksichtigt.
- Eine Bewertung möglicher Risiken, die sich aus der Literaturrecherche ergeben, wird oft nicht vorgenommen.
Leistungsbewertungsbericht (PER)
- Die im PER enthaltenen Daten werden nicht mit den spezifischen Anforderungen verknüpft, die damit erfüllt werden sollen.
- Eine klare Zusammenfassung der Ergebnisse, die darlegt, warum das Produkt dem Stand der Technik entspricht, insbesondere im Vergleich mit anderen Produkten, fehlt.
Häufige Mängel bei der Einreichung von Technischen Dokumentationen für IVDs
UNSER DOKUMENT ZUM DOWNLOAD
FAQ
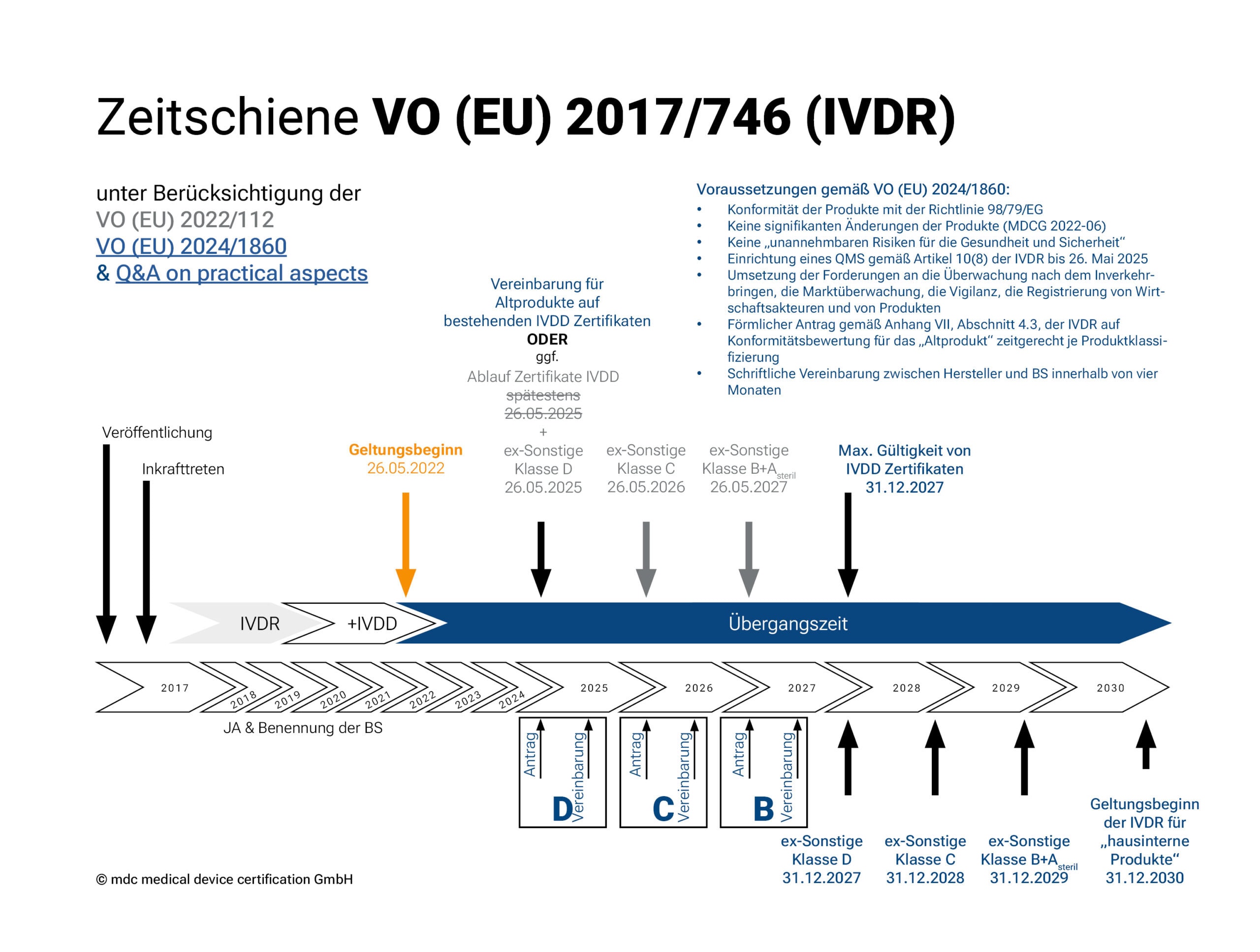
Übergangsfristen der IVDR gemäß Verordnung (EU) 2022/112
Der Geltungsbeginn der IVDR war der 26. Mai 2022. Im Dezember 2021 verlängerte die EU die Übergangsfristen der Verordnung 2017/746 über In-vitro-Diagnostika (s. 2022/112). Diese Verlängerung verschafft den Herstellern und Benannten Stellen mehr Zeit, um bereits im Verkehr befindliche IVD-Produkte durch das Konformitätsbewertungsverfahren zu bringen.
Je nach Risikoklassifizierung gelten (außer für Klasse A Produkte) verschiedene Übergangsfristen für die Umsetzung der neuen Verordnung. Bis 2027 (2028 für in-Haus Tests) soll die Verordnung vollständig umgesetzt sein.
- Klasse-A-Produkte (steril) erhalten zusammen mit Klasse-B-Produkten die längste Übergangsfrist für das Inverkehrbringen: bis 26. Mai 2027.
- Für Klasse-C-Produkte endet die Frist am 26. Mai 2026 und
- für Klasse-D-Produkte endet die Frist am 26. Mai 2025.
- Die sogenannte “Abverkaufsregelung” (Bereitstellung und Inbetriebnahme) entfällt.
Übergangsfristen der IVDR gemäß Verordnung (EU) 2024/1860
Die von der EU-Kommission veröffentlichte Anpassung der Übergangsfristen der IVDR in Form der Verordnung (EU) 2024/1860 ist zum 9. Juli 2024 in Kraft getreten und soll den Herstellern und den Benannten Stellen mehr Zeit zur Umsetzung der Vorgaben der IVDR gewähren. Die verlängerten Übergangsfristen gelten ausschließlich für Bestandsprodukte („legacy devices“) mit bestehender Konformitätsbescheinigung bzw. -erklärung und sind erneut nach der Risikoklasse des Bestandprodukts gestaffelt:
- Risikoklasse D und Produkte, die ein gültiges Zertifikat nach Richtlinie 98/79/EG besitzen: bis 31. Dezember 2027
- Risikoklasse C: bis 31. Dezember 2028
- Risikoklasse B und sterile Produkte der Risikoklasse A: bis 31. Dezember 2029
Die Verlängerung ist dabei an folgende Bedingungen geknüpft:
1. Diese Produkte entsprechen weiterhin der Richtlinie 98/79/EG;
2. Keine signifikanten Änderungen der Produkte (gemäß MDCG 2022-06);
3. Die Produkte stellen kein unvertretbares Risiko für die Gesundheit oder Sicherheit von Patienten, Anwendern oder anderen Personen oder für andere Aspekte des Gesundheitsschutzes dar;
4. Der Hersteller hat spätestens bis zum 26. Mai 2025 ein Qualitätsmanagementsystem gemäß Artikel 10 Absatz 8 der IVDR eingerichtet;
5. Der Hersteller hat spätestens zu folgenden Terminen einen formellen Antrag bei einer Benannten Stelle auf Konformitätsbewertung eingereicht und vier Monate später zur Unterzeichnung gebracht:
I. 26. Mai 2025 für Produkte der Risikoklasse D und Produkte, die ein gültiges Zertifikat nach Richtlinie 98/79/EG besitzen
II. 26. Mai 2026 für Produkte der Risikoklasse C
III. 26. Mai 2027 für Produkte der Risikoklasse B
Die Produkte, die durch ein Zertifikat gemäß der Richtlinie 98/79/EG abgedeckt sind, unterliegen in der Zeit weiterhin der Überwachung durch die Benannte Stelle.
Unabhängig von diesen Verlängerungen gelten zudem weiterhin bereits für die Bestandsprodukte die Anforderungen der IVDR zur Überwachung nach dem Inverkehrbringen, der Marktüberwachung, der Vigilanz und der Registrierung der Produkte und der Wirtschaftsakteure.
Zur Umsetzung hat die EU Kommission ein Q&A on practical aspects parallel veröffentlicht.
Was regelt die IVDR (EU) 2017/746
Die IVDR (EU) 2017/746 regelt die Anforderungen für das Inverkehrbringen, Bereitstellen und die Inbetriebnahme von IVD. Die Verordnung ist für die gesamte EU gültig und ersetzt die vorherige Richtlinie 98/79/EG.
Die Verordnung unterscheidet sich maßgeblich durch ein neues regelbasiertes Klassifizierungssystem. Im Rahmen der Konformitätsbewertung wird für wesentlich mehr In-vitro Diagnostika die Beteiligung einer Benannten Stelle gefordert. Ohne Beteiligung einer Benannten Stelle können nur noch nicht-sterile Produkte der Klasse A in Verkehr gebracht werden. Des Weiteren wurden die Anforderungen an das Vigilanzsystem und die Überwachung neu und detaillierter formuliert.
Durch die Verordnung soll eine transparente und neutrale Bewertung garantiert werden. Die eindeutige Produktidentifikation und Rückverfolgbarkeit von Produkten soll durch die Einführung der UDI (Unique Device Identifier) vereinfacht werden. Für mehr Transparenz und einen besseren Informationsfluss sorgt auch die europäische Datenbank (EUDAMED).
Als eine der wenigen Benannte Stellen Europas nach der IVDR (EU) 2017/746 Verordnung unterstützen wir Sie bei der Zertifizierung Ihrer In-vitro-Diagnostika (IVD) und begleiten Sie bei Ihrem Konformitätsbewertungsverfahren für IVD.
Vorteile der IVDR (EU) 2017/746
Die neue Verordnung bringt im Vergleich zu der vorherigen Richtlinie (IVDD) zahlreiche Änderungen, aber auch Vorteile für Patienten und Anwender von In-vitro Diagnostika, mit sich.
- Mehr Patientensicherheit durch hohe Standards für die Qualität und Produktsicherheit von In-vitro-Diagnostika
- Erhöhte Haftungs- uns Rechtssicherheit durch detailliertere Beschreibung der Anforderungen an die Produktsicherheit
- schnelles und effizientes Handeln bei Vorkommnissen / Rückrufen durch verbesserte Rückverfolgbarkeit mittels UDI (Unique Device Identification)
- Höhere Transparenz durch Datenspeicherung in der europäischen Datenbank (EUDAMED)
Für wen gilt die IVDR (EU) 2017/746?
Die EU VO 2017/746 gilt für alle, die In-vitro Diagnostika innerhalb der Europäischen Union in Verkehr bringen, auf dem Markt bereitstellen oder in Betrieb nehmen wollen.
Die EU-Verordnung 2017/746 IVDR fordert, dass In-Vitro Diagnostika bestimmten Risikoklassen (A bis D) zugeordnet werden. Diese regelbasierte Risikoklassifizierung (Anhang VIII) ersetzt die listenbasierte Kategorisierung unter der IVDD.
Die Beteiligung der Benannten Stelle an Konformitätsbewertungsverfahren ergibt sich aus der Produktklassifizierung nach Anhang VIII der Verordnung.
Die wichtigsten Änderungen der IVDR im Vergleich zur IVDD
Die EU-Medizinprodukteverordnung IVDR ersetzt die vorher gültige Richtlinie 98/79/EG IVDD.
Die neue Verordnung ist in 10 Kapitel und 15 Anhänge aufgeteilt und enthält 113 Artikel. Im Vergleich enthielt die IVDD 24 Artikel. Daraus wird ersichtlich, dass die IVDR deutlich umfangreicher ausfällt als die IVDD.
Das hat unter anderem zur Folge, dass die Zahl an IVD-Produkten, die eine Beteiligung der Benannten Stelle erfordern, von 7% unter der IVDD auf rund 80% unter der IVDR gestiegen ist.
- Verstärkte Einbeziehung der Benannten Stellen (Notified Bodies)
- Neues Klassifizierungssystem: Klassifizierung von IVD-Produkten in 4 Risikoklassen:
- Klasse A: Geringe Risiken für Patienten und die öffentliche Gesundheit.
- Klasse B: Mäßiges individuelles Risiko und/oder geringes Risiko für die öffentliche Gesundheit.
- Klasse C: Hohes individuelles Risiko und/oder mäßiges Risiko für die öffentliche Gesundheit.
- Klasse D: Hohes individuelles Risiko und hohes Risiko für die öffentliche Gesundheit.
Diese Klassifizierung ersetzt die listenbasierte Kategorisierung in der IVDD und hat einen großen Einfluss auf das Konformitätsverfahren, Zertifizierungsaudits und die Überwachung nach dem In-Verkehrbringen. Produkte ab der Klasse A steril oder höher benötigen unter der IVDR die Einbeziehung einer Benannten Stelle.
- Nachweis der klinischen Evidenz von Produkten (auf Basis des Performance-Evaluierungsberichts)
- Verschärfte Anforderungen an das Qualitätsmanagementsystem, die technische Dokumentation, das Vigilanzsystem und an die Überwachung nach dem Inverkehrbringen (Post Market Surveillance)
- Zuweisung von Pflichten der Wirtschaftsakteure (Hersteller, Bevollmächtigte, Importeure und Händler)
- Neue Kennzeichnungspflichten: Einführung einer neuen Produktidentifizierungsnummer (Unique Device Identification (UDI) für eine bessere Produktidentifikation bzw. Nachverfolgbarkeit von Produkten
- Erhöhte Transparenz durch Veröffentlichung von Daten über das Unternehmen sowie über Produkte und UDIs in der neuen Europäischen Datenbank für Medizinprodukte (EUDAMED). Die EUDAMED-Datenbank fungiert als Vigilanz- und Marktüberwachungssystem.
Nicht geändert hat sich, dass Hersteller weiterhin mittels eines Konformitätsbewertungsverfahrens nachweisen müssen, dass Ihre Prozesse und Ihre Produkte die nötigen Anforderungen erfüllen. Wie unter der IVDD muss auch unter der IVDR weiterhin die Konformität anhand einer CE-Kennzeichnung ausgedrückt werden.
Jetzt zu unserem Newsletter anmelden
KEINE NEUIGKEITEN MEHR VERPASSEN
Aktuelle Events
MDC VERANSTALTUNGEN
Unser LinkedIn-Auftritt
NEUIGKEITEN
Offene Stellenangebote
KARRIERE BEI MDC