

CE marking
Regulation (EU) 2017/746
The IVDR regulation is the applicable regulatory basis for in-vitro diagnostics (IVD) on the European market. In order to meet society’s ever-increasing demands on the safety and reliability of IVDs, Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) came into force in 2017, replacing the previous EU Directive 98/79/EC (IVDD). The transition period ended on 25 May 2022, but extended transition periods were adopted for class A sterile, B, C and D products. As one of the few Notified Bodies in Europe, mdc medical device certification GmbH is your competent partner for CE certification in accordance with the new regulations (EU) 2017/746.
Directly to the contact person
Any questions about the product? Click here to go directly to your contact person.
Services for the CE certification of in-vitro diagnostics
For manufacturers of in-vitro diagnostics, we perform the certifications required for CE marking in accordance with Regulation (EU) 2017/746 as a Notified Body. Conformity assessment procedures with Notified Bodies are mandatory for all manufacturers of class A (sterile) and class B, C and D devices. Our services also include the certification of patient-related tests and products for self-test by lay persons in the home environment.
The focus of our activities in this field is both the auditing of the QM system at the manufacturer’s sites and its subcontractors and the review of the technical documentation for the respective products in accordance with Annex IX. In addition to auditors with product and technology experience, we assign qualified technical experts with many years of expertise in industry, research institutes, laboratories and notified bodies. For the mandatory testing of manufactured products, we support the manufacturer in co-operation with notified EU reference laboratories.
| Classification | Assessment procedures offered by mdc |
|---|---|
| Class A (sterile) | Annex IX, Chapter I (the involvement of the Notified Body is limited to sterility aspects) |
| Class B | Annex IX, Chapters I and II This also includes the assessment of the technical documentation of at least one representative device per device category in accordance with Annex IX, Section 4 |
| Class B for self-testing or near-patient testing | In addition, the technical documentation of each device is assessed for the specific aspects according to section 5.1 |
| Class C | Annex IX, Chapters I and II This also includes the assessment of the technical documentation (of at least one representative product per generic product group) in accordance with Annex IX Section 4 |
| Class C for self-administration or near-patient testing | Includes an assessment of the technical documentation of each product for the specific aspects according to section 5.1 |
| Class D | Annex IX, Chapters I and II This also includes the assessment of the technical documentation for each product in accordance with Annex IX Section 4 (incl. verification by an EU reference laboratory) |
| Class D for self-testing or near-patient testing | Annex IX, Chapters I and II This also includes the assessment of the technical documentation according to Annex IX, Section 4 (incl. verification by an EU reference laboratory) and the specific requirements according to Section 5.1 |
Our certification is recognised by our cooperation partners for product authorisation in Ukraine.
Services for third countries
Product authorisation in Ukraine
We offer those interested in certifications the opportunity for an initial exchange of information.
CONTACT OUR IVDR TEAM
Procedure of a certification process
Here you can see an example of the processing of an mdc certification procedure in the field of in-vitro diagnostics as part of the CE marking required certifications according to Regulation (EU) 2017/746.
IVDR Documents
Overview of mdc documents on IVDR that may be of interest
01
Preparation
- Enquiry
- Preliminary review and offer
- Contract
- Application
02
Assessment
- Review of technical documentation (incl. SSP)
- Special procedures (e.g. for innovative products)
- Review of QM documents
- On-site audit (2-stage)
- Assessment report
03
Certification
- Evaluation of all activities and reports
- Decision
- Issue of certificate
04
Surveillance
- Review of technical documentation (random samples)
- Safety reports (PSUR)
- Surveillance audits
- Review of manufactured products (class D only)
- Assessment reports
- Processing of change notifications
- Unannounced audits
05
Re-Certification
- Preparation
- Assessment
- Certification
Additional services that may be of interest
EN ISO 13485
Here you can find more information about the product EN ISO 13485
Third countries
2017/745 (MDR)
Here you can find more information about the product 2017/745 (MDR)
Common Issues in the Submission of Technical Documentation for In Vitro Diagnostic Devices
The following points summarize frequently observed deficiencies in technical documentation submissions for IVDs. These examples are intended to highlight potential pitfalls and do not constitute formal guidance or recommendations.
Data Completeness and File Submission Errors
- Files are submitted in incorrect formats, such as *.docx files or ZIP files containing unnecessary subfolders (refer to the “Requirements for Submissions in Digital Format”, can be accessed via the service portal)
- File name length restrictions are not observed.
- Mandatory content required by Annexes II and III is incomplete or missing.
- Points marked as “not applicable” are not or insufficiently justified.
- Evidence of compliance is not clearly assigned or explicitly referenced (e.g., Unique Device Identifiers (UDI), supplier details, Certificates of Analysis (CoA)).
- Product samples for self-testing or near-patient testing devices are not provided.
General Content Deficiencies
- Instructions for Use (IFU) are incorrectly submitted as evidence for the product description, sampling procedures, or sample processing methods.
- The product description lacks critical information, including:
• Visual representations of the product (e.g., photos or drawings),
• Components and accessories (as required by the IVDR),
• Details on combination use (e.g., sample preparation, methods, devices, software),
• Market release format (e.g., kit, retail packaging, shipping packaging),
• The countries where the product will be marketed, including language requirements for labeling,
• Consistent descriptions of product variants across documents. - UDI information is insufficiently defined or unclear for individual products.
- The intended use does not comply with IVDR Annex I, Section 20.4.1(c), or is presented inconsistently across documents (e.g., Declaration of Conformity, product description, risk analysis, or performance evaluation). Missing indications and contraindications are common.
- The classification of the product as an IVD, as well as the corresponding risk class rule, is not sufficiently justified.
- Raw materials are misclassified, and specifications or packaging materials are incomplete or missing.
- Automated processes (e.g., instruments, software) are not adequately specified, including missing version numbers or revision details.
- Previous product generations or performance data from already marketed products are not clearly described or traceable.
- Labeling and IFUs are incomplete or missing in required languages, and residual risks are not adequately disclosed (e.g., warnings or contraindications).
- The development and manufacturing processes are not clearly documented, particularly outsourced processes and quality controls.
- General Safety and Performance Requirements (GSPR) (Annex I, IVDR) are not fully or transparently justified, and supporting evidence is often inadequately labeled.
- Gaps in risk management are evident, including unclear responsibilities, lack of clinical expertise, and insufficient risk assessments, especially for legacy products.
- Stability testing is poorly documented, with missing test plans or methodologies.
- The use of human, animal, or microbial materials is not sufficiently documented, and related safety evidence is incomplete.
- Post-Market Surveillance (PMS) findings are not integrated into the documentation, and the selection of comparable products or databases for PMS activities is not justified.
Performance Evaluation Deficiencies
A common issue in technical documentation submissions is the absence of a comprehensive and structured performance evaluation plan, particularly for “legacy products” already on the market. Manufacturers often fail to demonstrate how product conformity will be established. Key details regarding the use of historical data, literature sources, interlaboratory study results, or unpublished internal data are missing. When equivalent product data is used, the equivalence is not adequately justified.
Performance Evaluation Plan
- Justifications for non-applicable analytical and clinical performance characteristics are insufficient or unclear.
- Acceptance criteria for analytical and clinical studies are undefined, or references to relevant standards are missing.
- Statistical rationales for the number of samples or replicates and the selection of users or study groups are inadequately documented. References to applicable standards (e.g., Annexes, tables) are absent.
- Criteria for the acceptability of the risk-benefit ratio are not defined.
Analytical Performance
- Evidence is often based on legacy devices without clearly addressing differences between the legacy product and the product under evaluation. The transferability of legacy data to the IVDR-compliant product is insufficiently justified
- Study design descriptions are incomplete, lacking details on sample materials, acceptance criteria, sample size rationale, and methodology. References to internal SOPs or external guidelines (e.g., CLSI) are insufficient.
- Definitions for outliers and exclusion criteria are missing, and repeat measurements are not addressed.
- Justifications for comparator products and the selection of interfering substances and cross-reactants are absent.
- Combination use, such as integrating multiple devices or methods, is inadequately documented.
Clinical Performance Evaluation
- Qualifications of principal investigators for clinical performance studies are not adequately presented.
- Comparability between the legacy product and the device under evaluation is not sufficiently established.
- Definitions of outliers and exclusion criteria, along with measures for repeat testing, are missing.
- Rationale for not collecting clinical data in user environments or clinical settings is absent.
- Study reports are not signed by an authorized individual or physician. Information on study location, duration, and materials used is often incomplete.
- Results are frequently summarized superficially, without clearly linking individual study outcomes to the test reports.
- Deviations or excluded data are insufficiently discussed or explained.
Data from Literature and Other Sources
- Literature search protocols are incomplete, missing search terms, inclusion/exclusion criteria, or key words.
- The state of the art is insufficiently supported or documented.
- When clinical performance is based solely on literature, a detailed summary of both positive and negative findings is absent.
- Risks identified through literature review are not assessed or discussed.
Performance Evaluation Report (PER)
- Data in the PER is not explicitly linked to specific requirements.
- A clear summary explaining how the product meets the state of the art, particularly compared to other products, is missing.
Common deficiencies in the submission of technical documentation for IVDs
DOWNLOAD OUR DOCUMENT
FAQ
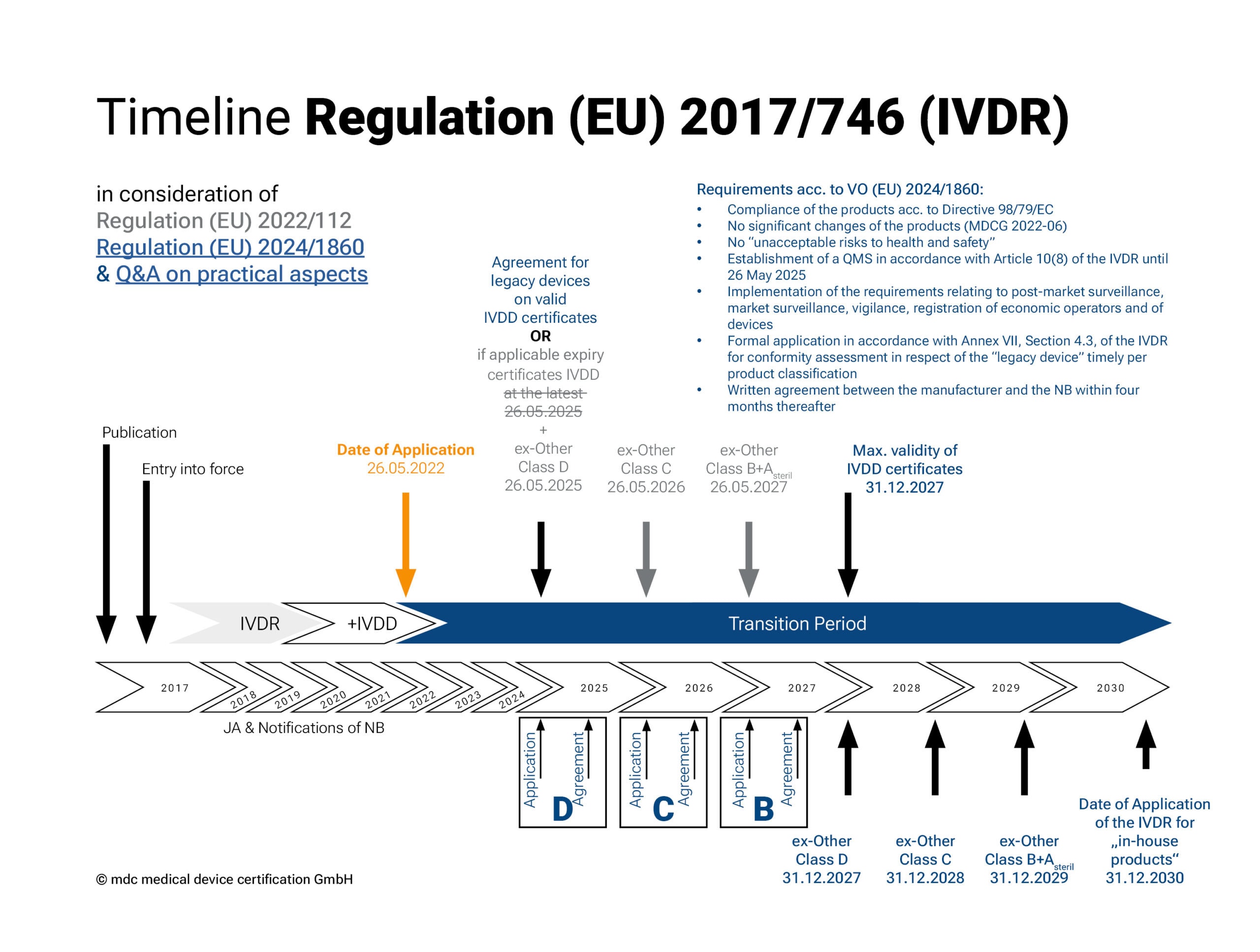
Transitional periods of the IVDR in accordance with Regulation (EU) 2022/112
The IVDR came into force on 26 May 2022. In December 2021, the EU extended the transitional periods of Regulation 2017/746 on in vitro diagnostic medical devices (see 2022/112). This extension gives manufacturers and notified bodies more time to bring IVD products already on the market through the conformity assessment procedure.
Depending on the risk classification (except for Class A devices), different transition periods apply for the implementation of the new regulation. The regulation should be fully implemented by 2027 (2028 for in-house tests).
• Class A products (sterile), together with Class B products, have the longest transitional period for placing on the market: until 26 May 2027.
• For Class C products, the deadline is 26 May 2026 and
• for Class D products, the deadline is 26 May 2025.
• The so-called “sell-off regulation” (provision and commissioning) no longer applies.
Transitional periods of the IVDR in accordance with Regulation (EU) 2024/1860
The adaptation of the transitional periods for the IVDR, published by the EU Commission in the form of Regulation (EU) 2024/1860, came into force on 9. July 2024 and is intended to give manufacturers and notified bodies more time to implement the requirements of the IVDR. The extended transitional periods apply exclusively to legacy devices with an existing conformity certificate or declaration of conformity and are again staggered according to the risk class of the legacy device:
• Risk class D and devices with a valid certificate under Directive 98/79/EC until 31 December 2027
• Risk class C: until 31 December 2028
• Risk class B and sterile devices of risk class A: until 31 Dezember 2029
The extension is subject to the following conditions:
1. These devices continue to comply with Directive 98/79/EC;
2. No significant changes have been made to the devices (according to MDCG 2022-06);
3. The devices do not pose an unacceptable risk to the health or safety of patients, users, or other persons, or to other aspects of health protection;
4. The manufacturer has established a quality management system according to Article 10(8) of the IVDR by 26 May 2025;
5. The manufacturer has submitted a formal application for conformity assessment to a notified body and has completed it by the following deadlines:
I. 26 May 2025 for risk class D devices and devices with a valid certificate under Directive 98/79/EC
II. 26 May 2026 for risk class C devices
III. 26 May 2027 for risk class B devices
Devices covered by a certificate according to Directive 98/79/EC will continue to be monitored by the notified body during this period. Regardless of these extensions, the requirements of the IVDR for post-market surveillance, market surveillance, vigilance, and the registration of devices and economic operators still apply to legacy devices.
The EU Commission has published a Q&A on practical aspects in parallel.
What does the IVDR Regulation (EU) 2017/746 regulate?
The IVDR (EU) 2017/746 regulates the requirements for the placing on the market, making available and putting into service of IVDs. The regulation applies to the entire EU and replaces the previous Directive 98/79/EC.
The regulation is characterised by a new rule-based classification system. As part of the conformity assessment, the involvement of a notified body is required for significantly more in-vitro diagnostics. Without the involvement of a notified body, only non-sterile class A devices can be placed on the market. Furthermore, the requirements for the vigilance system and monitoring have been reformulated and made more detailed.
The regulation is intended to guarantee a transparent and neutral assessment. The introduction of the UDI (Unique Device Identifier) is intended to simplify the unambiguous identification and traceability of products. The European database (EUDAMED) also ensures greater transparency and a better flow of information.
As one of the few Notified Bodies in Europe under the IVDR (EU) 2017/746 Regulation, we support you in the certification of your in-vitro diagnostics (IVD) and assist you with your conformity assessment procedure for IVDs.
Advantages of the IVDR (EU) 2017/746
Compared to the previous directive (IVDD), the new regulation brings with it numerous changes, but also advantages for patients and users of in-vitro diagnostics.
• Greater patient safety thanks to high standards for the quality and product safety of in-vitro diagnostics
• Increased liability and legal certainty thanks to a more detailed description of product safety requirements
• Fast and efficient action in the event of incidents / recalls thanks to improved traceability using UDI (Unique Device Identification)
• Greater transparency through data storage in the European database (EUDAMED)
Who does the IVDR (EU) 2017/746 apply to?
EU Regulation 2017/746 applies to all those who place in-vitro diagnostics on the market, make them available on the market or wish to put them into service within the European Union.
EU Regulation 2017/746 IVDR requires that in-vitro diagnostics are assigned to certain risk classes (A to D). This rule-based risk classification (Annex VIII) replaces the list-based categorisation under the IVDD.
The involvement of the notified body in conformity assessment procedures results from the product classification according to Annex VIII of the regulation.
What are the differences between the IVDR and IVDD
The EU Medical Device Regulation IVDR replaces the previously valid Directive 98/79/EC IVDD.
The new regulation is divided into 10 chapters and 15 annexes and contains 113 articles. In comparison, the IVDD contained 24 articles. This shows that the IVDR is significantly more comprehensive than the IVDD.
One of the consequences of this is that the number of IVD products requiring the involvement of the notified body has risen from 7% under the IVDD to around 80% under the IVDR.
- Increased involvement of notified bodies
- New classification system: Classification of IVD products into 4 risk classes:
- Class A: Low risk to patients and public health.
- Class B: Moderate individual risk and/or low risk to public health.
- Class C: High individual risk and/or moderate risk to public health.
- Class D: High individual risk and high risk to public health.
This classification replaces the list-based categorisation in the IVDD and has a major impact on the conformity procedure, certification audits and post-market surveillance. Devices from class A sterile or higher require the involvement of a Notified Body under the IVDR.
- Proof of the clinical evidence of devices (based on the performance evaluation report)
- Stricter requirements for the quality management system, technical documentation, vigilance system and post-market surveillance
- Assignment of obligations to economic operators (manufacturers, authorised representatives, importers and distributors)
- New labelling obligations: Introduction of a new product identification number (Unique Device Identification (UDI)) for better product identification and traceability of products
- Increased transparency through publication of data on the company, products and UDIs in the new European Database for Medical Devices (EUDAMED). The EUDAMED database acts as a vigilance and market surveillance system.
Not changed has the obligation for manufacturers to proof that their processes and products fulfil the necessary requirements by means of a conformity assessment procedure. As under the IVDD, the conformity under the IVDR must be expressed by the CE marking.
Subscribe our newsletter
DON’T MISS ANY MORE NEWS
Upcoming events
EVENTS BY MDC
Our LinkedIn profile
NEWS
Open vacancies
CAREER AT MDC