
Notification of non-production periods or production times
Designation_MDR
List of Medical Devices (MDR)
Notification of Change (MDR)
Price List – Certification according to Regulation (EU) 2017/745 (MDR) & Recognition in Third Countries
Basic UDI-DI and its assignment
The Basic UDI-DI is an important key in product documentation (e.g. certificates, technical documentation, vigilance notifications and PSUR, SS(C)P, etc.) and also serves as an access key to the product-related information in the European database for medical devices (EUDAMED). The Basic UDI-DI is a useful way of grouping several similar variants of a medical device.
Essential requirements for defining the Basic UDI-DI are listed in MDCG 2018-1 (currently Rev. 4). Only MDR requirements are addressed, but the guidance document is considered applicable for Regulation (EU) 2017/746 as well.These requirements must be taken into account:
The following data elements must be the same for all devices that are grouped together.
Requirements for grouping under the same Basic UDI-DI:
- Same manufacturer (name, address, SRN)
- Same risk class (in particular with regarding implantability, active medical device, special components such as drug component, materials of animal origin or substances, but also measuring function or whether it is a reusable surgical element)
- Same Medical Device Nomenclature Code (EMDN Code) – at least up to the fourth digit
- Same intended purpose
- Listed on the same product certificate, PSUR, SSCP (MP) or SSP (IVD)
- Same technical documentation (if applicable)
- Same essential design and manufacturing characteristics (if applicable)
For IVD, one Basic UDI-DI is assigned in particular for language and distribution variants (e.g. tests for self-testing in different countries for distributors with country-specific labelling) as well as packaging variants of the same product (e.g. 96-well or 128-well plates, multiple packaging).
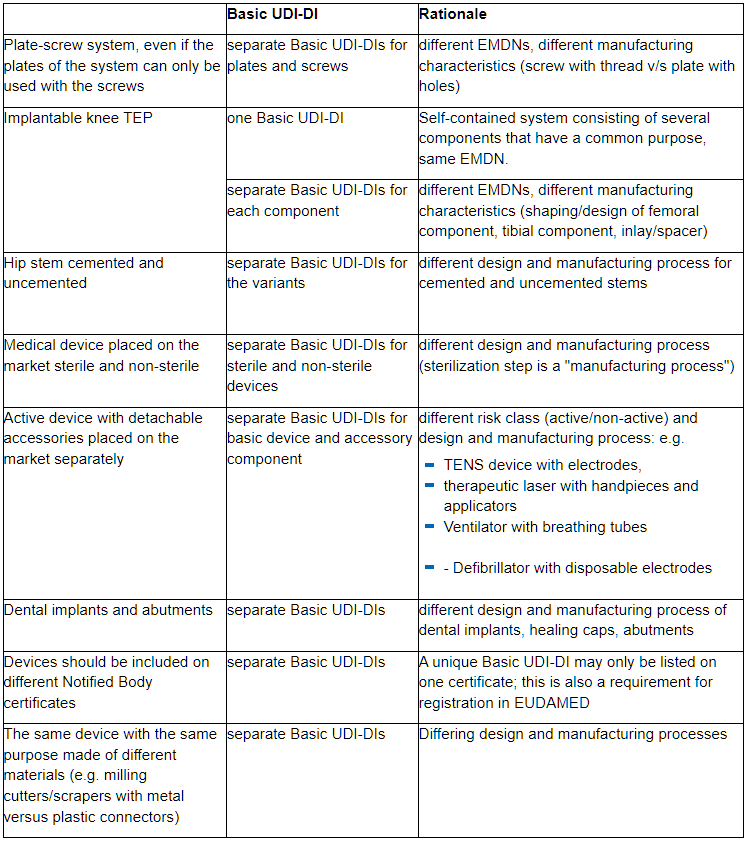
In particular with regard to the same essential design and manufacturing characteristics, we would like to point out some aspects that typically lead to different Basic UDI-DIs for medical devices:
Binding provision of the German market surveillance authorities (AGMP) about the handling of declarations of conformity for legacy devices (according to 93/42/EEC and 98/79/EC)
At present, there is no need to request a current declaration of conformity, if there are no changes, in addition to the original declaration of conformity issued before the date of application of the MDR (26 May 2021) or IVDR (26 May 2022), with reference to Art. 120 MDR or Article 110 IVDR.
However, in the event of non-significant changes that require an amendment to the declaration of conformity (e.g. change of name, change of address), the existing declaration of conformity is supplemented by an addendum or annex in accordance with the directive.
This has now been clarified by the German market surveillance authorities (AGMP) and it is particularly pointed out that there may NOT be a new declaration of conformity according to MDD/ IVDD, but only a “supplement” (see above) to the declaration of conformity issued before 2021-05-26 (MDD)/ 2022-05-26 (IVDD), which clearly states the non-significant changes (e.g. also new product variants).
Binding provision of the German market surveillance authorities (AGMP) about the handling of declarations of conformity for legacy devices (according to 93/42/EEC and 98/79/EC)
At present, there is no need to request a current declaration of conformity, if there are no changes, in addition to the original declaration of conformity issued before the date of application of the MDR (26 May 2021) or IVDR (26 May 2022), with reference to Art. 120 MDR or Article 110 IVDR.
However, in the event of non-significant changes that require an amendment to the declaration of conformity (e.g. change of name, change of address), the existing declaration of conformity is supplemented by an addendum or annex in accordance with the directive.
This has now been clarified by the German market surveillance authorities (AGMP) and it is particularly pointed out that there may NOT be a new declaration of conformity according to MDD/ IVDD, but only a “supplement” (see above) to the declaration of conformity issued before 2021-05-26 (MDD)/ 2022-05-26 (IVDD), which clearly states the non-significant changes (e.g. also new product variants).
ISO 13485
ISO 9001
Downloads
Designation_MDR
List of Medical Devices (MDR)
Technical Documentation (Medical Devices) – File Structure (ZIP)
Notification of Change (MDR)
Price List – Certification according to Regulation (EU) 2017/745 (MDR) & Recognition in Third Countries
Client-News
Extension of the Transitional Period for the IVDR in Accordance with Regulation (EU) 2024/1860
The adaptation of the transitional periods for the IVDR, published by the EU Commission in the form of Regulation (EU) 2024/1860, came into force on 9. July 2024 and is intended to give manufacturers and notified bodies more time to implement the requirements of the IVDR. The extended transitional periods apply exclusively to legacy devices with an existing conformity certificate or declaration of conformity and are again staggered according to the risk class of the legacy device:
• Risk class D and devices with a valid certificate under Directive 98/79/EC until 31 December 2027
• Risk class C: until 31 December 2028
• Risk class B and sterile devices of risk class A: until 31 Dezember 2029
The extension is subject to the following conditions:
1. These devices continue to comply with Directive 98/79/EC;
2. No significant changes have been made to the devices (according to MDCG 2022-06);
3. The devices do not pose an unacceptable risk to the health or safety of patients, users, or other persons, or to other aspects of health protection;
4. The manufacturer has established a quality management system according to Article 10(8) of the IVDR by 26 May 2025;
5. The manufacturer has submitted a formal application for conformity assessment to a notified body and has completed it by the following deadlines:
I. 26 May 2025 for risk class D devices
II. 26 May 2026 for risk class C devices
III. 26 May 2027 for risk class B devices
IV. fore the expiry of the certificate under Directive 98/79/EC for the relevant devices
Devices covered by a certificate according to Directive 98/79/EC will continue to be monitored by the notified body during this period. Regardless of these extensions, the requirements of the IVDR for post-market surveillance, market surveillance, vigilance, and the registration of devices and economic operators still apply to legacy devices.
The EU Commission has published a Q&A on practical aspects in parallel.
Basic UDI-DI and its assignment
The Basic UDI-DI is an important key in product documentation (e.g. certificates, technical documentation, vigilance notifications and PSUR, SS(C)P, etc.) and also serves as an access key to the product-related information in the European database for medical devices (EUDAMED). The Basic UDI-DI is a useful way of grouping several similar variants of a medical device.
Essential requirements for defining the Basic UDI-DI are listed in MDCG 2018-1 (currently Rev. 4). Only MDR requirements are addressed, but the guidance document is considered applicable for Regulation (EU) 2017/746 as well.These requirements must be taken into account:
The following data elements must be the same for all devices that are grouped together.
Requirements for grouping under the same Basic UDI-DI:
- Same manufacturer (name, address, SRN)
- Same risk class (in particular with regarding implantability, active medical device, special components such as drug component, materials of animal origin or substances, but also measuring function or whether it is a reusable surgical element)
- Same Medical Device Nomenclature Code (EMDN Code) – at least up to the fourth digit
- Same intended purpose
- Listed on the same product certificate, PSUR, SSCP (MP) or SSP (IVD)
- Same technical documentation (if applicable)
- Same essential design and manufacturing characteristics (if applicable)
For IVD, one Basic UDI-DI is assigned in particular for language and distribution variants (e.g. tests for self-testing in different countries for distributors with country-specific labelling) as well as packaging variants of the same product (e.g. 96-well or 128-well plates, multiple packaging).
In particular with regard to the same essential design and manufacturing characteristics, we would like to point out some aspects that typically lead to different Basic UDI-DIs for medical devices:
Binding provision of the German market surveillance authorities (AGMP) about the handling of declarations of conformity for legacy devices (according to 93/42/EEC and 98/79/EC)
At present, there is no need to request a current declaration of conformity, if there are no changes, in addition to the original declaration of conformity issued before the date of application of the MDR (26 May 2021) or IVDR (26 May 2022), with reference to Art. 120 MDR or Article 110 IVDR.
However, in the event of non-significant changes that require an amendment to the declaration of conformity (e.g. change of name, change of address), the existing declaration of conformity is supplemented by an addendum or annex in accordance with the directive.
This has now been clarified by the German market surveillance authorities (AGMP) and it is particularly pointed out that there may NOT be a new declaration of conformity according to MDD/ IVDD, but only a “supplement” (see above) to the declaration of conformity issued before 2021-05-26 (MDD)/ 2022-05-26 (IVDD), which clearly states the non-significant changes (e.g. also new product variants).
Binding provision of the German market surveillance authorities (AGMP) about the handling of declarations of conformity for legacy devices (according to 93/42/EEC and 98/79/EC)
At present, there is no need to request a current declaration of conformity, if there are no changes, in addition to the original declaration of conformity issued before the date of application of the MDR (26 May 2021) or IVDR (26 May 2022), with reference to Art. 120 MDR or Article 110 IVDR.
However, in the event of non-significant changes that require an amendment to the declaration of conformity (e.g. change of name, change of address), the existing declaration of conformity is supplemented by an addendum or annex in accordance with the directive.
This has now been clarified by the German market surveillance authorities (AGMP) and it is particularly pointed out that there may NOT be a new declaration of conformity according to MDD/ IVDD, but only a “supplement” (see above) to the declaration of conformity issued before 2021-05-26 (MDD)/ 2022-05-26 (IVDD), which clearly states the non-significant changes (e.g. also new product variants).
PSUR for MDR-/ IVDR-certified devices
Reminder for the necessity of the submission of Periodic Safety Update Reports.
With a certain frequency manufacturers of medical devices of risk classes IIa, IIb and III have to create a PSUR (Periodic Safety Update Report) according to MDR Art. 86 and manufacturers of In-vitro-Diagnostics of risk classes C and D have to create a PSUR according to IVDR Art. 81. For MDR-/ IVDR-certified devices these PSURs have to be submitted to the Notified Body and will be assessed by it according to the requirements.
MDCG 2022-21 contains comprehensive explanations regarding the content of the PSUR for medical devices.
For MDR-/ IVDR-certified devices for which mdc did not receive a PSUR yet (in consideration of the period of creation), mdc will contact the manufacturers to remind the necessity and deadlines.
Timelines for Transition of Certification from MDD to MDR
As of March 20, 2023, Regulation (EU) 2023/607 was published to smoothen the transition from MDD (and AIMDD) to MDR certification of devices on the market and to assure availability of medical devices on the European market. Manufacturers of devices certified according to Medical Device Directive (MDD), also known as “legacy devices” according to MDR Article 120, can now benefit from longer periods for the transition of MDD certification to MDR. In order to be able to benefit from this extension let’s delve into the key highlights:
Extension of MDR validity of certificates:
All certificates not expired on March 20, 2023 have been extended by definition until December 31, 2027 (for class III and class IIb implantable legacy devices) and until December 31, 2028 (for other class IIb and class IIa, as well as class Im and Is legacy devices).
It has to be noted that an additional prerequisite for placing devices on the market has been introduced: additional to the valid MDD certificate the manufacturer must have applied for the individual device (or a successor device) for MDR certification by May 26, 2024.
The notified body can then issue a confirmation letter (as defined in the Q&A document by the European Commission) confirming formally the validity of the MDD certificate.
However, it’s important to note that the extension is subject to certain conditions to benefiting from the new transition period. Manufacturers must:
- by May 26, 2024 establish a quality management system (QMS) compliant with MDR;
- by May 26, 2024 submit a formal application for conformity assessment under MDR;
- by September 26, 2024 have a mutually signed written agreement with the Notified Body for conformity assessment under MDR.
Additionally, the extension of the transitional regime only applies to MDR legacy devices that:
- continue to comply with Directive 93/42/EEC;
- do not undergo significant changes in design or intended use;
- do not pose unacceptable risks to the health and safety of patients and users.
The new regulation also introduces additional requirements for legacy devices with expired certificates. Also such devices may benefit from the extended transition period either if the manufacturer had concluded a contract with a Notified Body for conformity assessment under the MDR before the expiration of the MDD certificate or has obtained exemptions under Article 97 or 59 MDR.
This extension of the transition period addresses the demands of the medical device industry, which has long sought a more flexible certification deadline for MDD legacy devices. It is good news for medical device manufacturers. However, it is crucial to act promptly and meet the first deadline of May 26, 2024, to ensure compliance with the new rules.
In this context we may also remind on the revision of MDCG 2020-3 (Rev. 1) on the definition of significant changes. The document gives clarification on the conditions when a change can be considers as “non-significant” and thus be implemented under an existing MDD certificate. This is especially crucial to adopt devices in the next four to five years to maintain safety and availability in the transition period.
Additional transition periods for Custom made devices:
Implantable class III custom made devices need to have a quality management certification according to Annex IX, by May 26, 2026 in order to continue to place these devices on the market.
Removal of the “sell-off” period:
The new regulation eliminates the previously defined “sell-off” period stated in MDR, Article 120(4).
Additional transition periods for Annex XVI devices:
Also the timelines for implementation of MDR for products without an intended medical purpose (Annex XVI, MDR) have recently been extended. By Commission Implementing Regulation (EU) 2022/2346 the legal framework, i.e. the Common Specifications, for certification of Annex XVI devices was generally established. The original version was also amended to align the transition periods with definitions of Regulation (EU) 2023/607 – this was published on June 20 2023 as Commission Implementing Regulation (EU) 2023/1194. It was clarified that if the device was MDD certified timelines of Art. 120 MDR (see above) shall apply. In all other cases (device not MDD certified) also the timeline for transition of the devices to MDR certification are extended.
Prerequisites:
- Device was lawfully marketed in the Union before 22 June 2023
- in continuous accordance with applicable requirements and
- not substantially changed.
Applicable Timelines for Annex-XVI-devices the manufacturer intends to perform or is performing a clinical investigation (CI):
- 22 June 2024: confirmation by the National Competent Authority acc. to Art. 70 (1) or (3) MDR that the application for the CI is complete;
- 23 December 2024: the sponsor must have started the CI;
- 1 January 2028: written agreement for the conformity assessment has been signed with the Notified Body must be in place;
- 31 December 2029: end of transition period.
Applicable Timelines for Annex-XVI-devices the manufacturer does not perform a clinical investigation:
- 1 January 2027: written agreement for the conformity assessment has been signed with the Notified Body must be in place
- 31 December 2028: end of transition period
Extension transitionperiodes for MDR
On 20 Mar. 2023, the European Commission published Regulation (EU) 2023/607 extending the transitional periods for devices requiring a Notified Body for conformity assessment under Regulation (EU) 2017/745 (MDR).
UPDATE: On July 2023 the European Commission has additionally published a updated Q&A document on the extension of the timelines. The information can be found on the EC website:
The new regulation newly regulates the validity of AIMDD and MDD certificates and defines requirements for the first placing on the market of “legacy devices” (as per MDCG 2021-25), even if the validity of the underlying certificates according to the directive (MDD or AIMDD) has formally expired.
Overview of the changes:
- Extension of the transitional periods for the first placing on the market of devices based on certificates according to MDD or AIMDD, for all devices for which an application for certification (see below) according to MDR was submitted before the expiry of the MDD certificate until 31 Dec. 2027 (Class III and implantable devices Class IIb [with exceptions]) or until 31 Dec. 2028 (all other devices).
- Extension of the transition period for class I devices (MDD) that are classified higher under MDR until Dec. 31, 2028.
- Extension of transition period for Class III custom-made devices until Dec. 31, 2027.
For these devices (except custom-made devices), the manufacturer must have established a QM system according to MDR by 26 May 2024 at the latest. The second, essential prerequisite for being able to benefit from the extended timelines is a written contract between the manufacturer and the notified body for certification according to MDR (certification contract). This contract must be applied for at the latest before the expiry of the MDD certificate and must cover all products for which the extension of the transitional periods is to be claimed. The basis of the contract is the availability of the technical documentation in accordance with the requirements of the MDR.
In order to be able to submit an application for certification – either initial MDR certification or extension of the MDR certification for new device groups – we require a questionnaire (initial certification) or an amended list of medical devices (a notification of change alone is not sufficient). On this basis, we will provide you with an offer for certification. This offer is the basis for your application for certification and subsequently for the final certification contract. We will implement the requirements for the contract in such a way that, together with the application for certification, at least one complete technical documentation must be submitted for review in addition to the documentation of the QM system. The technical documentation according to MDR requirements for the other products covered by the certification contract must also be available at the time of application, but can be submitted for review at a later date according to an agreed plan.
In order to be able to issue the confirmation letter proposed by the EU Commission, a contract for the monitoring of the (formally expired) MDD certificates must also be concluded. On the basis of this communication this confirmation letter can be issued confirming that products may continue to be placed on the market with the CE mark and mdc’s identification number 0483. We are preparing the corresponding application procedure and will make them available as soon as possible.
Alternatively, the manufacturer (or his European authorized representative) can apply for an exemption according to Art. 97 MDR at the responsible market surveillance authority, this is independent of the status of the MDD certificates.
Another significant change is also the elimination of the sell-by period for medical devices and In- vitro Diagnostic Devices placed on the market under AIMDD, MDD and IVDD.
You can also find the text of the regulation at
https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32023R0607
Products without a medical purpose – Anh. XVI procedure
As of Dec. 1, 2022, the European Commission published the definitions of common specifications for the devices without a medical purpose listed in Annex XVI of the MDR (Regulation (EU) 2022/2346. The implementing regulation contains, in particular, specifications for the conformity assessment procedure and the requirements to be taken into account – so-called common specifications.
In particular, specific requirements and transition periods are defined for all those products that were previously not subject to medical device law but must meet the requirements of the MDR. The basic requirement is that the product has already been lawfully placed on the Union market before 22 June 2023 and continues to meet the legal requirements. Furthermore, the design and intended purpose of the product must not undergo any significant changes ( as defined in Art. 120 (3) MDR and MDCG 2020-3).According to Article 2 (2) of Regulation (EU) 2022/2346, these products may be placed on the market or put into service from September 22, 2023, until June 22, 2025, if a written agreement on the performance of the conformity assessment has been signed between the Notified Body and the manufacturer.
Special deadlines have also been set for devices for which a clinical investigation is carried out as part of the initial conformity assessment as a medical device.
Furthermore, we would like to refer to the current European discussion regarding the transition periods for already MDD-certified products, which may also affect products regulated under Annex XVI MDR.
We will inform our existing customers about the possibilities and procedures at mdc in time to meet these deadlines.
Surveillance of legacy devices under Article 120 of the MDR (MDCG 2022-4) and Article 110 (3) of the IVDR (MDCG 2022-15)
With the position papers MDCG 2022-4 and MDCG 2022-15, Notified Bodies are requested to identify relevant certificates (under Directive 93/42/EEC and 98/79/EC) which are subject to surveillance according to Article 120 (3) of the MDR or Article 110 (3) of the IVDR. The requirement for adequate surveillance of existing legacy devices1/2 by Notified Bodies is sharpened and should be reflected in the audit. This means that the following aspects will become a focus of the audit:
- Transition strategy to MDR / IVDR
- Assessment of (non-) significant changes according to MDCG 2020-3 or MDCG 2022-6
- Adjustments of the requirements according to Article 120 (3) of the MDR or Article 110 (3) of the IVDR in the QMS (including post-market surveillance (Annex III of the MDR / IVDR), market surveillance, vigilance and registration of economic operators and devices)
- Assess whether all appropriate processes related to post-market surveillance, including risk management and performance data, are included in the post-market surveillance plan
For manufacturers, in addition to maintaining the QM system, all requirements must be implemented or mapped.
1Products placed on the market under Article 120(3) of the MDR after the date of application of the MDR (26 May 2021) and until the end of the transitional period on 26 May 2024, provided that certain conditions are met.
2Products placed on the market under Article 110(3) of the IVDR after the date of application of the IVDR (26 May 2022) and until the end of the respective transitional period referred to in the second or third subparagraph of Article 110(3), provided that certain conditions are met.
MDR – EU Commission asks manufacturers for applications
In the new position paper MDCG 2022-11, which has been published by the European Commission, manufacturers are requested to submit their applications for certification under MDR early and completely.
Herewith MDCG considers the fact that according to an information by TEAM-NB, the European association of Notified Bodies for medical devices, a majority of the members reported that more than half of the applications have to be considered as incomplete, which means that the quality management documentation or the technical documentation is not in status, which allows an assessment.
It is mentioned that Notified Bodies may not be in a position to perform an assessment of all technical documentations until May 2024. Further the document clarifies that potential exceptional approvals by national Competent Authorities can only be performed if the products concerned are needed in the interest of public health, patient safety of patient health.
mdc fully supports this call for timely applications, which are complete and compliant based on the experience with the MDR made so far. Complete submissions should be made to mdc already in 2022 in order to maintain a chance for a certification until May 2024.