
Häufige Fehler bei der Einreichung von Technischen Akten (IVDR)
Auf unseren Infoveranstaltungen im Februar zum Übergang von der IVDD zur IVDR haben wir intensiv über häufige Fehler bei der Einreichung technischer Dokumentationen gesprochen. Viele Unternehmen stehen vor ähnlichen Herausforderungen – daher haben wir als Benannte Stelle die häufigsten Abweichungen nun auch auf unserer Website veröffentlicht.
Was erwartet Sie?
- Eine Übersicht typischer Fehler bei der Einreichung technischer Akten
- Praktische Hinweise zur Vermeidung häufiger Fallstricke
- Alle Infos als gebündeltes Dokument zum Download
Meldung über produktionsfreie Zeiten oder Produktionszeiten
Infoveranstaltung ‚DER LANGE WEG VON IVDD ZU IVDR‘ in Stuttgart
Diese Woche fand die erste unserer beiden Infoveranstaltungen zum Übergang von der IVDD zur IVDR statt – und wir freuen uns über einen gelungenen Austausch!
Gemeinsam mit Experten und Teilnehmern haben wir zentrale Fragen zur IVDR-Transition diskutiert:
- Welche Schritte sind (dringend) notwendig?
- Welche Fristen und Regularien sind zu beachten?
- Wie gelingt eine IVDR-konforme technische Dokumentation?
Besonders wertvoll war der direkte Austausch mit unseren Auditoren und Fachexperten – sei es in den Vorträgen oder in der offenen Fragerunde. Vielen Dank an alle Teilnehmenden für das große Interesse, die spannenden Diskussionen und wertvollen Einblicke!
Wir freuen uns darauf, den Dialog fortzusetzen.
Start (Copy)
Verlängerung der Übergangszeit der IVDR gemäß Verordnung (EU) 2024/1860
Die von der EU-Kommission veröffentlichte Anpassung der Übergangsfristen der IVDR in Form der Verordnung (EU) 2024/1860 ist zum 9. Juli 2024 in Kraft getreten und soll den Herstellern und den Benannten Stellen mehr Zeit zur Umsetzung der Vorgaben der IVDR gewähren. Die verlängerten Übergangsfristen gelten ausschließlich für Bestandsprodukte („legacy devices“) mit bestehender Konformitätsbescheinigung bzw. -erklärung und sind erneut nach der Risikoklasse des Bestandprodukts gestaffelt:
• Risikoklasse D und Produkte, die ein gültiges Zertifikat nach Richtlinie 98/79/EG besitzen: bis 31. Dezember 2027
• Risikoklasse C: bis 31. Dezember 2028
• Risikoklasse B und sterile Produkte der Risikoklasse A: bis 31. Dezember 2029
Die Verlängerung ist dabei an folgende Bedingungen geknüpft:
1. Diese Produkte entsprechen weiterhin der Richtlinie 98/79/EG;
2. Keine signifikanten Änderungen der Produkte (gemäß MDCG 2022-06);
3. Die Produkte stellen kein unvertretbares Risiko für die Gesundheit oder Sicherheit von Patienten, Anwendern oder anderen Personen oder für andere Aspekte des Gesundheitsschutzes dar;
4. Der Hersteller hat spätestens bis zum 26. Mai 2025 ein Qualitätsmanagementsystem gemäß Artikel 10 Absatz 8 der IVDR eingerichtet;
5. Der Hersteller hat spätestens zu folgenden Terminen einen formellen Antrag bei einer Benannten Stelle auf Konformitätsbewertung eingereicht und vier Monate später zur Unterzeichnung gebracht:
I. 26. Mai 2025 für Produkte der Risikoklasse D
II. 26. Mai 2026 für Produkte der Risikoklasse C
III. 26. Mai 2027 für Produkte der Risikoklasse B
IV. vor Ablauf des Zertifikat nach Richtlinie 98/79/EG für betreffende Produkte
Die Produkte, die durch ein Zertifikat gemäß der Richtlinie 98/79/EG abgedeckt sind, unterliegen in der Zeit weiterhin der Überwachung durch die Benannte Stelle.
Unabhängig von diesen Verlängerungen gelten zudem weiterhin bereits für die Bestandsprodukte die Anforderungen der IVDR zur Überwachung nach dem Inverkehrbringen, der Marktüberwachung, der Vigilanz und der Registrierung der Produkte und der Wirtschaftsakteure.
Zur Umsetzung hat die EU Kommission ein Q&A on practical aspects parallel veröffentlicht.
Benennungsurkunde_IVDR
Preisliste – Zertifizierung nach Verordnung (EU) 2017/746 (IVDR)
Fragebogen zur Angebotserstellung – In-vitro Diagnostika – Anlage: Liste In-vitro-Diagnostika (IVDR)
Änderungsmitteilung (IVDR)
Basis UDI-DI und deren Vergabe
Die Basis-UDI-DI ist wichtiger Schlüssel in der Produkt-Dokumentation (z. B. Zertifikate, technische Dokumentation, Vigilanz Meldungen und PSUR, SS(C)P, etc.) und dient auch als Zugangsschlüssel zu den produktbezogenen Informationen in der europäischen Datenbank für Medizinprodukte (EUDAMED). Die Basis-UDI-DI stellt dabei eine sinnvolle Möglichkeit dar, mehrere vergleichbare Varianten eines Medizinprodukts zu gruppieren.
Wesentliche Anforderungen für die Festlegung der Basis-UDI-DI sind in MDCG 2018-1 (aktuell Rev. 4) angeführt. Es werden nur Forderungen der 2017/745 (MDR) adressiert, das Dokument wird aber auch für die Verordnung (EU) 2017/746 herangezogen. Diese Anforderungen müssen berücksichtigt werden.
Insbesondere bei der gemeinsamen Gruppierung von Produkten sind wesentliche Aspekte zu berücksichtigen. Die folgenden Datenelemente müssen für alle gemeinsam gruppierten Produkte gleich sein.
Anforderungen für das Zusammenfassen unter derselben Basis-UDI-DI:
- Selber Hersteller (Name, Adresse, SRN)
- Selbe Risikoklasse (MDR: insbesondere hinsichtlich Implantierbarkeit, aktives Medizinprodukt, besondere Bestandteile wie Arzneimittelkomponente, Materialien tierischen Ursprungs oder stoffliche Komponenten sowie Messfunktion oder ob es sich um ein wiederverwendbares chirurgisches Element handelt; IVDR: gemäß Anhang VIII)
- Selber Medical Device Nomenclature Code (EMDN Code) – zumindest bis zur vierten Stelle
- Selbe Zweckbestimmung
- Angeführt auf demselben Produktzertifikat, PSUR, SSCP (MP) bzw. SSP (IVD)
- Gegebenenfalls gleiche (eine) Technische Dokumentation
- Gegebenenfalls s gleiche wesentliche Auslegungs- und Herstellungsmerkmale
Für IVD wird eine Basis-UDI-DI insbesondere für Sprach- und Vertriebsvarianten (z.B. Tests zur Eigenanwendung in verschiedenen Ländern für diverse Distributoren mit länderspezifischer Kennzeichnung) sowie Packungsvarianten des gleichen Produkts (z.B. 96-Well bzw. 128-Well Platten, Mehrfachabpackungen) vergeben.
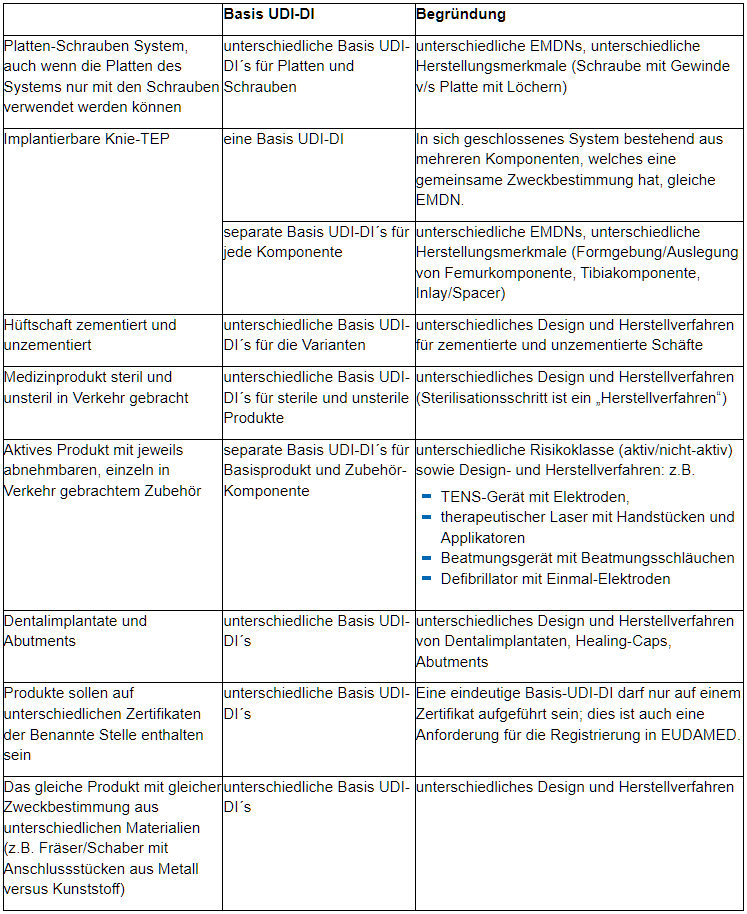
Insbesondere hinsichtlich derselben wesentlichen Auslegungs- und Herstellungsmerkmale möchten wir für Medizinprodukte auf einige Aspekte hinweisen, welche zu unterschiedlichen Basis-UDI-DI führen:
Downloads
Benennungsurkunde_IVDR
Preisliste – Zertifizierung nach Verordnung (EU) 2017/746 (IVDR)
Fragebogen zur Angebotserstellung – In-vitro Diagnostika – Anlage: Liste In-vitro-Diagnostika (IVDR)
Änderungsmitteilung (IVDR)
Kunden News
Häufige Fehler bei der Einreichung von Technischen Akten (IVDR)
Auf unseren Infoveranstaltungen im Februar zum Übergang von der IVDD zur IVDR haben wir intensiv über häufige Fehler bei der Einreichung technischer Dokumentationen gesprochen. Viele Unternehmen stehen vor ähnlichen Herausforderungen – daher haben wir als Benannte Stelle die häufigsten Abweichungen nun auch auf unserer Website veröffentlicht.
Was erwartet Sie?
- Eine Übersicht typischer Fehler bei der Einreichung technischer Akten
- Praktische Hinweise zur Vermeidung häufiger Fallstricke
- Alle Infos als gebündeltes Dokument zum Download
Infoveranstaltung ‚DER LANGE WEG VON IVDD ZU IVDR‘ in Stuttgart
Diese Woche fand die erste unserer beiden Infoveranstaltungen zum Übergang von der IVDD zur IVDR statt – und wir freuen uns über einen gelungenen Austausch!
Gemeinsam mit Experten und Teilnehmern haben wir zentrale Fragen zur IVDR-Transition diskutiert:
- Welche Schritte sind (dringend) notwendig?
- Welche Fristen und Regularien sind zu beachten?
- Wie gelingt eine IVDR-konforme technische Dokumentation?
Besonders wertvoll war der direkte Austausch mit unseren Auditoren und Fachexperten – sei es in den Vorträgen oder in der offenen Fragerunde. Vielen Dank an alle Teilnehmenden für das große Interesse, die spannenden Diskussionen und wertvollen Einblicke!
Wir freuen uns darauf, den Dialog fortzusetzen.
Verlängerung der Übergangszeit der IVDR gemäß Verordnung (EU) 2024/1860
Die von der EU-Kommission veröffentlichte Anpassung der Übergangsfristen der IVDR in Form der Verordnung (EU) 2024/1860 ist zum 9. Juli 2024 in Kraft getreten und soll den Herstellern und den Benannten Stellen mehr Zeit zur Umsetzung der Vorgaben der IVDR gewähren. Die verlängerten Übergangsfristen gelten ausschließlich für Bestandsprodukte („legacy devices“) mit bestehender Konformitätsbescheinigung bzw. -erklärung und sind erneut nach der Risikoklasse des Bestandprodukts gestaffelt:
• Risikoklasse D und Produkte, die ein gültiges Zertifikat nach Richtlinie 98/79/EG besitzen: bis 31. Dezember 2027
• Risikoklasse C: bis 31. Dezember 2028
• Risikoklasse B und sterile Produkte der Risikoklasse A: bis 31. Dezember 2029
Die Verlängerung ist dabei an folgende Bedingungen geknüpft:
1. Diese Produkte entsprechen weiterhin der Richtlinie 98/79/EG;
2. Keine signifikanten Änderungen der Produkte (gemäß MDCG 2022-06);
3. Die Produkte stellen kein unvertretbares Risiko für die Gesundheit oder Sicherheit von Patienten, Anwendern oder anderen Personen oder für andere Aspekte des Gesundheitsschutzes dar;
4. Der Hersteller hat spätestens bis zum 26. Mai 2025 ein Qualitätsmanagementsystem gemäß Artikel 10 Absatz 8 der IVDR eingerichtet;
5. Der Hersteller hat spätestens zu folgenden Terminen einen formellen Antrag bei einer Benannten Stelle auf Konformitätsbewertung eingereicht und vier Monate später zur Unterzeichnung gebracht:
I. 26. Mai 2025 für Produkte der Risikoklasse D
II. 26. Mai 2026 für Produkte der Risikoklasse C
III. 26. Mai 2027 für Produkte der Risikoklasse B
IV. vor Ablauf des Zertifikat nach Richtlinie 98/79/EG für betreffende Produkte
Die Produkte, die durch ein Zertifikat gemäß der Richtlinie 98/79/EG abgedeckt sind, unterliegen in der Zeit weiterhin der Überwachung durch die Benannte Stelle.
Unabhängig von diesen Verlängerungen gelten zudem weiterhin bereits für die Bestandsprodukte die Anforderungen der IVDR zur Überwachung nach dem Inverkehrbringen, der Marktüberwachung, der Vigilanz und der Registrierung der Produkte und der Wirtschaftsakteure.
Zur Umsetzung hat die EU Kommission ein Q&A on practical aspects parallel veröffentlicht.
Basis UDI-DI und deren Vergabe
Die Basis-UDI-DI ist wichtiger Schlüssel in der Produkt-Dokumentation (z. B. Zertifikate, technische Dokumentation, Vigilanz Meldungen und PSUR, SS(C)P, etc.) und dient auch als Zugangsschlüssel zu den produktbezogenen Informationen in der europäischen Datenbank für Medizinprodukte (EUDAMED). Die Basis-UDI-DI stellt dabei eine sinnvolle Möglichkeit dar, mehrere vergleichbare Varianten eines Medizinprodukts zu gruppieren.
Wesentliche Anforderungen für die Festlegung der Basis-UDI-DI sind in MDCG 2018-1 (aktuell Rev. 4) angeführt. Es werden nur Forderungen der 2017/745 (MDR) adressiert, das Dokument wird aber auch für die Verordnung (EU) 2017/746 herangezogen. Diese Anforderungen müssen berücksichtigt werden.
Insbesondere bei der gemeinsamen Gruppierung von Produkten sind wesentliche Aspekte zu berücksichtigen. Die folgenden Datenelemente müssen für alle gemeinsam gruppierten Produkte gleich sein.
Anforderungen für das Zusammenfassen unter derselben Basis-UDI-DI:
- Selber Hersteller (Name, Adresse, SRN)
- Selbe Risikoklasse (MDR: insbesondere hinsichtlich Implantierbarkeit, aktives Medizinprodukt, besondere Bestandteile wie Arzneimittelkomponente, Materialien tierischen Ursprungs oder stoffliche Komponenten sowie Messfunktion oder ob es sich um ein wiederverwendbares chirurgisches Element handelt; IVDR: gemäß Anhang VIII)
- Selber Medical Device Nomenclature Code (EMDN Code) – zumindest bis zur vierten Stelle
- Selbe Zweckbestimmung
- Angeführt auf demselben Produktzertifikat, PSUR, SSCP (MP) bzw. SSP (IVD)
- Gegebenenfalls gleiche (eine) Technische Dokumentation
- Gegebenenfalls s gleiche wesentliche Auslegungs- und Herstellungsmerkmale
Für IVD wird eine Basis-UDI-DI insbesondere für Sprach- und Vertriebsvarianten (z.B. Tests zur Eigenanwendung in verschiedenen Ländern für diverse Distributoren mit länderspezifischer Kennzeichnung) sowie Packungsvarianten des gleichen Produkts (z.B. 96-Well bzw. 128-Well Platten, Mehrfachabpackungen) vergeben.
Insbesondere hinsichtlich derselben wesentlichen Auslegungs- und Herstellungsmerkmale möchten wir für Medizinprodukte auf einige Aspekte hinweisen, welche zu unterschiedlichen Basis-UDI-DI führen:
Verbindliche Festlegung der deutschen Marktaufsichtsbehörden (AGMP) zum Umgang mit Konformitätserklärungen für Richtlinien-Produkte (gemäß 93/42/EWG bzw. 98/79/EG)
Zur Zeit besteht keine Notwendigkeit eine aktuelle Konformitätserklärung, wenn es keine Änderungen gibt, ergänzend zur ursprünglichen, vor dem Geltungsbeginn der MDR (26.05.2021) bzw. IVDR (26.05.2022) ausgestellten Konformitätserklärung, mit Bezug auf Art. 120 MDR bzw. Artikel 110 IVDR zu fordern.
Jedoch muss im Falle nicht signifikanter Änderungen, die eine Anpassung der Konformitätserklärung erfordern (z.B. Umfirmierung, Adressänderung) die bestehende Konformitätserklärung gemäß Richtlinie um ein Beiblatt bzw. Anhang ergänzt werden.
Dies wurde nun durch die deutschen Marktaufsichtsbehörden (AGMP) klargestellt und es wird besonders daraufhin gewiesen, dass es KEINE neue Konformitätserklärung nach MDD/ IVDD geben darf, sondern nur ein „Beiblatt“ (s.o.) zu der vor dem 2021-05-26 (MDD)/ 2022-05-26 (IVDD) ausgestellten Konformitätserklärung, welche die nicht signifikanten Änderungen (wie auch neue Produktvarianten) eindeutig nennt.
Erstes Zertifikat für Produkte unter der IVDR ausgestellt
Wir freuen uns, die Übergabe des ersten Zertifikats für Produkte unter der Verordnung (EU) 2017/746 (IVDR) an die Firma MIKROGEN GmbH bekannt zu geben.
Harald Rentschler gratulierte MIKROGEN und würdigte die Übergabe des Zertifikates als einen wichtigen Meilenstein sowohl für den Hersteller als auch die Benannte Stelle. Er betonte, dass die umfangreichen Anforderungen der IVDR für beide Seiten sehr große Herausforderungen bedeuten, welche nicht nur fachlich sondern auch administrativ zu bewältigen waren.
Sein Dank ging sowohl an die Geschäftsleitung und die für die Umsetzung der IVDR verantwortlichen Personen von MIKROGEN als auch an die zuständige Projektleitung und die gutachterlich Tätigen der mdc. Das Zertifikat stelle einen erneuten Beweis für eine gelungene, mittlerweile mehr als 20-jährige Zusammenarbeit der beiden Firmen dar.
PSUR / Sicherheitsberichte für MDR-/ IVDR-zertifizierten Produkten
Erinnerung an die Notwendigkeit zur Einreichung von Sicherheitsberichten
Hersteller von Medizinprodukten der Risikoklassen IIa, IIb und III müssen gemäß MDR Art. 86 bzw. Hersteller von In-vitro-Diagnostika der Risikoklassen C und D müssen gemäß IVDR Art. 81 in einer bestimmten Frequenz Sicherheitsberichte erstellen. Für MDR-/ IVDR-zertifizierte Produkte sind diese Sicherheitsberichte bei der Benannten Stelle einzureichen und werden von ihr gemäß der Anforderungen geprüft.
Das MDCG 2022-21 enthält umfassende Erläuterungen zum Inhalt des Sicherheitsberichts für Medizinprodukte.
Für die MDR-/ IVDR-zertifizierten Produkte, für die mdc (unter Berücksichtigung der Erstellungsfristen) noch keine Sicherheitsberichte erhalten hat, wird mdc auf die Kunden zukommen und an die Notwendigkeit und Fristen erinnern.
Benennung unter der IVDR (EU) 2017/746 Verordnung
Mit der Veröffentlichung in der europäischen Datenbank NANDO ist die offizielle Liste der Benannten Stellen unter der Verordnung (EU) 2017/746 (IVDR) heute mit der mdc medical device certification GmbH um eine weitere Position gewachsen.
Mit der heutigen Veröffentlichung in der europäischen Datenbank NANDO, haben wir als Benannte Stelle die offizielle Benennung unter der Verordnung (EU) 2017/746 (IVDR) erhalten. Die 2017 verabschiedete Verordnung gilt in der gesamten EU und ersetzt die vormalige Richtlinie 98/79/EG.Nach einer mehrjährigen und aufwendigen Vorbereitungs-, Begutachtungs- und Genehmigungszeit sind wir sehr stolz darauf, diesen für uns und unsere Kunden wichtigen Meilenstein erreicht zu haben. Die Geschäftsleitung der mdc bedankt sich herzlich bei allen Mitarbeiterinnen und Mitarbeitern, die zum Erreichen dieses Ziels in der Firmengeschichte beigetragen haben und bei allen Kunden für ihre Treue während der langen Antragsphase.
Wir freuen uns sehr darauf, Ihr kompetenter Partner im Rahmen der CE-Kennzeichnung Ihrer Produkte auch unter den neuen Regularien zu sein und stehen für entsprechende Anfragen gerne zur Verfügung.
Kontakt: ivd(at)mdc-ce.de
25.01.2022: EU ändert Übergangsbestimmungen der IVDR
Mit der Verordnung (EU) 2022/112 hat die EU die Übergangsbestimmungen für bestimmte In-vitro-Diagnostika geändert und den Geltungsbeginn der IVDR für „hausinterne Produkte“ bis zum 26. Mai 2028 aufgeschoben. Obgleich der Anwendungsbeginn am 26.05.2022 bestehen bleibt, erlaubt der neue Rechtsrahmen für viele In-vitro-Diagnostika, die sich bereits im Verkehr befinden, längere Übergangsfristen:
- Bescheinigungen, die von Benannten Stellen gemäß Richtlinie 98/79/EG ausgestellt wurden, verlieren erst spätestens am 27. Mai 2025 ihre Gültigkeit. Dies bedeutet, dass Produkte, welche unter der Richtlinie 98/79/EG durch eine Benannte Stelle zertifiziert wurden, bis zum 26. Mai 2025 in Verkehr gebracht oder in Betrieb genommen werden können.
- Altprodukte, welche vor dem 26.05.2022 über eine gültige Konformitätserklärung gemäß Richtlinie 98/79/EG verfügten und ohne das Mitwirken einer Benannten Stelle auf dem Markt bereitgestellt wurden, dürfen in Abhängigkeit von der Risikoklasse bis zu folgenden Zeitpunkten in Verkehr gebracht werden:
- 26. Mai 2025 für Produkte der Klasse D;
- 26. Mai 2026 für Produkte der Klasse C;
- 26. Mai 2027 für Produkte der Klasse B;
- 26. Mai 2027 für Produkte der Klasse A, die in sterilem Zustand in Verkehr gebracht werden.
Zusätzlich wurden Fristen für die Bereitstellung auf dem Markt und die Inbetriebnahme festgelegt.
Für das Änderungsmanagement der Produkte, die gemäß Richtlinie 98/79/EG in Verkehr gebracht oder in Betrieb genommen werden, erwarten wir analog zu MDCG 2020-3 (Guidance on significant changes regarding the transitional provision under Article 120 of the MDR with regard to devices covered by certificates according to MDD or AIMDD) ein ähnliches Guidance Dokument.
Die Anforderungen der IVDR an die Überwachung nach dem Inverkehrbringen, die Marktüberwachung, die Vigilanz sowie die Registrierung von Wirtschaftsakteuren und von Produkten gelten jedoch für alle IVD ab dem 26.05.2022.
25 Jahre mdc medical device certification GmbH
Die Gründung der Zertifizierungsgesellschaft mdc medical device certification GmbH wurde heute vor 25 Jahren, am 10. Dezember 1996 zur Eintragung beim Handelsregister Memmingen angemeldet.
Bereits 1994 war „mdc medical device certification“ die Bezeichnung eines Geschäftsbereichs der Dr. Müller-Lierheim GmbH, welcher mit der 1996 vorgenommenen Unternehmensgründung in die rechtliche Eigenständigkeit überführt wurde.
Seit der Fusion mit der Zertifizierungsstelle Medizinprodukte von ZDH-ZERT e.V. im Jahre 2000 ist der Sitz der mdc in Stuttgart. Weitere Bürostandorte befinden sich in Berlin, Tuttlingen und Wien. Ferner wird in wenigen Wochen in Haifa (Israel) die erste außereuropäische Niederlassung bezogen. In den vergangenen 25 Jahren ist die Anzahl der Angestellten von drei auf über 100 gestiegen. Zusätzlich sind ungefähr 70 freiberuflich tätige Auditoren, Fachexperten und Begeher tätig. Das Tochterunternehmen des ZDH-ZERT Verein für Qualität im Handwerk und in der gewerblichen Wirtschaft e.V. gehört nicht nur zu den führenden Benannten Stellen und Präqualifizierungsstellen sondern ist auch Marktführer bei der Zertifizierung von QM-Systemen in Betrieben der Gesundheitshandwerke und Hilfsmittelversorger.
Die mdc hat ihre Aktivitäten stets auf das Gebiet der Medizinprodukte und verwandte Bereiche fokussiert. Derzeit deckt das Spektrum die Tätigkeit als Benannte Stelle unter der Verordnung (EU) 2017/745 für Medizinprodukte (MDR) und unter der Richtlinie 98/79/EG für In-vitro Diagnostika (IVDD), als akkreditierte Zertifizierungsstelle für QM-Systeme gemäß ISO 13485 und ISO 9001 sowie als akkreditierte Präqualifizierungsstelle im Gebiet der Hilfsmittelversorgung ab. Abgerundet wird das Angebot durch Anerkennungen in der Ukraine und in Taiwan, durch Audits unter dem Medical Device Single Audit Program (MDSAP) im Rahmen eines Kooperationsverfahrens sowie die Veranstaltung öffentlicher Präsenz- und Onlineseminare zu Themen aus dem Bereich Qualitätsmanagement und Regulatory Affairs. Unter der Verordnung (EU) 2017/746 (IVDR) befindet sich mdc in einem fortgeschrittenen Stadium des Benennungsverfahrens.
Anlässlich des 25-jährigen Firmenjubiläums durfte der Geschäftsführer Harald Rentschler, der diese Position bei der Gründung antrat, von der Geschäftsführerin der IHK Stuttgart, Frau Dr. Susanne Herre eine Ehrenurkunde der IHK entgegen nehmen. Er dankt allen, die das Unternehmen auf seinem bisherigen erfolgreichen Weg begleitet haben.